R.Rotomskis,
V.Vaicaitis, D.Frolov, S.Bagdonas,
Laser Research Centre, Vilnius University, Sauletekio 9, 2040 Vilnius
Lithuania
![]()
R.Rotomskis,
V.Vaicaitis, D.Frolov, S.Bagdonas, |
|
|
| Discussion In some papers it was suggested that TPPS4 does not stack in water [26] since the negative charges at the sulfonato groups, cause the electrostatic repulsion between TPPS4 molecules. Our experimental results clearly support the assumption conclusion that the dimerization equilibrium exists for TPPS4 as well. On the basis of the model proposed below we would try to explain how the negative charges on the sulfonato groups could be involved in the formation of face-to-face dimers. Both the absorption and emission spectra of concentrated aqueous solutions of TPPS4 at pH7.5 showed evidence of strong interaction between the porphyrin rings. The increase of the TPPS4 concentration in a solution slightly shifts the Soret band to the blue and broadens it significantly, especially on the blue wing (Fig.4a). A shift of Q bands towards the longer wavelengths, which is most distinctive for the last band Qx(0,0), and redistribution of absorption intensity between two central bands are typical for the TPPS4 solutions at higher concentration in the visible spectral region. Theoretical considerations and experimental data concerning other tetrapyrrolic dimers have shown that the shifts of absorption bands associated with dimerization can be used to differentiate between the two following dimer models: a) side-by-side structure, in which the aggregates are formed by electrostatic interaction between side ligands, and b) a face-to-face structure with porphyrins stacked mostly by p -p interaction. A theoretical analysis of the effect of porphyrin dimerization on the absorption spectrum [27] predicts a negligible effect for the side-by-side dimer. In the case when linear dimers are formed the Soret band becomes wider and sometimes slightly shifted to the red [28-30]. On the contrary, in the case of face-to-face type dimers the Soret band is blue-shifted (800-1800 cm-1), its half-width increases [21] and the Q bands undergo a slight shift (100-300 cm-1) to the red part of spectrum [31]. Theoretically predicted spectroscopic features of dimers have been observed in the spectra of synthesized porphyrin with well-defined linear or stacked (face-to-face) structures [32-35]. Our spectroscopic data demonstrate that the linear structure cannot account for the observed changes in the spectra, whereas it could be satisfactorily explained by the formation of dimers with the stacked structure. Nevertheless, it is difficult to expect that the negatively charged sulfonato- groups of TPPS4 would tend to be located in close proximity in the face-to-face dimer, keeping in mind the fact that phenyl groups are nearly perpendicular to the plane of porphyrin ring in non-protonated species. Karns et al. [36] suggested a dimeric structure of haematoporphyrin consisting of two overlapping rings with negatively charged propanoate groups extended as far as possible from each other. Then the favourable structure for a face-to-face dimer of TPPS4 could be such where both steric constraints induced by out-of-plane sulfonatophenyl groups and electrostatic repulsion of negative charges localized on the sulfonato group are minimized by 45∞ rotation of one monomer in respect of the other. There is no exciton split for a stacked structure, where one molecule of TPPS4 is turned at the 45∞ angle so that dipole transitions of two monomers become nonparallel. Therefore the spectrum of the Soret band in this case must be almost identical to that of an isolated monomer [37], and only the broadening of the Soret band without any shift might be expected. However, our spectroscopic measurements show that in aqueous solutions of higher TPPS4 concentration the Soret band is slightly shifted to the blue. The observed changes in spectral characteristics of the Soret band are typical for those of face-to-face type aggregates when the transition dipoles are parallel or antiparallel [37]. This behaviour has been observed for a large number of face-to-face (free base) compounds and predicted by the molecular exciton theory [32, 38]. Similar spectral broadening and a red shift of the Q bands have been reported upon porphyrin aggregation [18] and formation of complexes with heterocycles [39]. Therefore, considering that the structure with nonparallel dipole transitions discussed above is not supported by our spectroscopic data, we propose another TPPS4 dimer model (Fig.11).
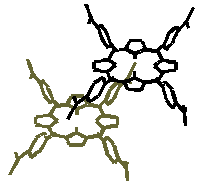
Fig.11 Structure of TPPS4 dimer. In this model the planes of porphyrins are shifted relatively to each other so that one of four sulfonato- groups of monomer is placed over/under the centre of macrocycle of other monomer. We suppose that purely excitonic (p -p ) interaction between porphyrin rings causes the shift of the Soret band to the blue [21, 33-35] and has an effect on the Q bands. The absorption of aggregated TPPS4 in the visible spectral region is very similar to that of covalently liked basket-type [38] or strati-bisporphyrins [32]. The reorganization of the Q bands might be slightly influenced by the weak incipient charge-transfer interactions between sulfonato group of one porphyrin molecule and nitrogens of other as well. Charges of the peripheral sulfonato- groups had a strong effect on the distribution of the electronic density near nitrogen atoms of a tetrapyrrolic macrocycle [18]. As a consequence, a charge transfer state may be formed due to interactions between negatively charged substitutes of one porphyrin monomer and the nitrogen in the center of other monomer. This would also lead to stronger van der Waals interaction for a face-to-face dimer. The detailed analysis of the spectroscopic features of TPPS4 and their relation to covalently linked and equilibrium structures supports the conclusion that in neutral aqueous solutions TPPS4 forms the face-to-face type aggregates (Fig.11). It is reasonable to expect that at the range of TPPS4 concentrations used for time-resolved spectroscopy (Fig.9) samples contained both face-to-face dimers as well as monomeric porphyrins. Obviously, the registered absorption changes must reflect excitation energy relaxation in different species present in aqueous solution (monomers and aggregates). However, the decay of transient absorption of TPPS4 cannot be fitted by a biexponential function in aqueous solution at neutral pH (Fig.10). The best approximation was achieved by single-exponential function (relaxation lifetime t around 3 ns). The dependence of excitation relaxation time on sample concentration and decrease in intensity of detected absorption changes with increasing concentration at the same intensity of pump pulse used led to the conclusion that excitation relaxation time in face-to-face aggregates might be very short. The changes observed in the absorption spectrum in aqueous solution of TPPS4 with the increase of the medium acidity (Fig.6) could be a result of the formation of the protonated species H2+P(SO3-)4 as it was reported earlier [12, 16, 18, 24]. The increase of porphyrin concentration in this case leads to the appearance of a new absorption peak at around 490 nm and a small increase of absorbance at around 710 nm for the (first time detected in [40]) indicating the formation of J-aggregates [15, 16, 23] from protonated species. It seems that the negative charges located on the side substitutes of dications play a crucial role in ensuring stability of the formed structures. The disruption of J-aggregates at the extreme pH values indicated by the disappearance of the characteristic peak (Fig.7) could be caused by the protonation of sulfonato- groups. The absorption kinetics of TPPS4 J-aggregates under excitation can be tentatively approximated with biexponential function having the time constants about t 1=27 ps and t 2=1.44 ns, or with three exponential decay function having the time constants t 1=24 ps, t 2= 290 ps and t 3=2-3 ns (Fig.10). Careful analysis of the fluorescence decay constants obtained in TPPS4 solutions for different concentrations at variuos pH values shows that the decay constant around 0.2 ns reflects the relaxation of excitation in J-aggregates. Decay constant values of 3.7 ns, 7 ns and 10 ns measured in the selected pH range from ñ1.1 to 7 can be attributed to the energy relaxation of dication (H2+P(SO3-)4), and non-protonated (P(SO3-)4) species of TPPS4, correspondingly. A short decay value of 0.5 ns detected in solutions at neutral pH reflects the formation of aggregated TPPS4 species, probably face to face dimers. Fluorescence decay rates were measured at fluorescence maxima for non-protonated protonated and aggregated TPPS4 and the results of fluorescence lifetime measurements were summarized in [16]. The fluorescence lifetimes of the non-protonated and protonated monomers of TPPS4 presented in [16, 26, 41] agree with each other quite well. However, for the J-aggregates there is a significant spread in fluorescence lifetimes obtained using different techniques such as phase modulation, streak camera and single photon counting [16]. The lifetimes of induced absorption in the J-aggregates obtained from experimental data (Fig.9c) approximately agree with two of the three components in the case when three-exponential approximation was applied (t 1=50ps; t 2=290ps; t 3=2.08ns, [41]) or with data were the major and second components of two-exponential decay have relaxation times of 82ps and 1.7ns [14, 16]. We suppose that the differences in the component of the shortest lifetime might be attributed to singlet-singlet annihilation process [42] since the intensive pump pulse was used in our experiments. An approximation of decay with exponential functions yields the values of process lifetimes which are usually applied to describe the systems consisting of monomers and small aggregates. However, in the case when molecules build large aggregates and excitation in such aggregate can be described by the features of the exciton the singlet-singlet annihilation becomes possible. Therefore, due to the very large size of aggregates pulses of very low energy should be used in order to obtain exact lifetime of the excitation relaxation. While another model bearing other specific parameters should be applied to describe relaxation processes in such systems more thoroughly, an exponential approximation giving excitation lifetimes is still commonly used for comparison purposes.
ACKNOWLEDGMENT The authors gratefully acknowledge financial support by the Lithuanian State Foundation for Science and Studies. |
![]()
![]()
|
|
|
![]()


{kind=link}
{kind=link}
{kind=link}