Using Fluorine-Induced Chemiluminescence To Detect Organo-Metalloids in the Headspace of Phototrophic Bacterial Cultures Amended with Selenium and TelluriumVerena Van Fleet-Stalder and Thomas G. ChasteenDepartment of Chemistry and Texas Regional Institute for Environmental Studies Sam Houston State University, Huntsville,Texas, USA
Brief Introduction of Chemiluminescence MethodsAnalytical methods based on chemiluminescence have taken their strong position among the more mundane analytical techniques because of a triumvirate of strengths: sensitivity, selectivity, and in many cases, a wide linear detection range. This is true even though chemiluminescence is not as widely applicable as absorption, emission, or even fluorescence methods of detection since so few molecules undergo chemiluminescent reactions. Because chemiluminescence, light emission generated from a chemical reaction, requires no light source for excitation, the analytical signal appears out of an essentially black background, and the only background signal is that of the photomultiplier tube's (PMT) dark current. Therefore light source warm-up and drift and interference from light scattering are absent. In the case of systems where red and near infrared light are observed in analytical detection, red sensitive PMT's dark current can be minimized by cooling; with blue light emission detection, cooling is not required. Detection limits routinely orders of magnitude lower than fluorescence methods are achievable. In addition, interfering molecules are often less of a problem too since chemiluminescence reactions can be so selective.The initial reports in the scientific literature of lophine (2,4,5-triphenyl imidazole) and then lucigenin (N,N'-dimethyl-9,9'-diacridinium nitrate) chemiluminescence in the last quarter of the nineteenth century [1] blossomed in this century into reports involving many different chemiluminescent reagents. The most common or well known solution phase systems involve luminol (or its derivatives), oxalate esters, lucigenin (or its derivatives), ruthenium tris-bipyridine, and luciferin. Gas phase examples include the ozone- and fluorine-induced, sodium vapor, and chlorine dioxide chemiluminescence detectors for gas chromatography. Because many of the solution phase systems use hydrogen peroxide or organic peroxides as oxidant and these can be generated many ways in liquid systems and because many of the solution phase reagents mentioned above can be tagged onto a large variety of analytes, high performance liquid chromatographic (HPLC) solution phase chemiluminescence is more common and variously applied than gas phase chemiluminescence reactions. In a very general way, the requirements for the analytically useful production of light from a chemical reaction are: 1) excess chemical energy produced by the reaction must be relatively efficiently used to populate the excited state of the emitter and 2) the excited species must have few mechanism of deactivation except light emission. In many systems, the initially excited state molecule is used as a conduit of energy to excite a second or third molecule which, instead, is the actually emitting species. In solution-phase systems, pH and catalysts must also be considered; however, in gas phase systems reaction cell pressure and temperature can be important factors. A more detailed description of these phenomenon and their applications can be found in the literature in a number of places [1, 2, 3, 4, 5, 6, 7, 8, 9]. The use of chemiluminescence as a detection method following analytical separation makes up a significant share of its application. For example, liquid phase chemiluminescence has been applied to high performance liquid chromatography [10, 11] and very recently to capillary electrophoresis [12, 13, 14]. Gas phase analytical chemiluminescence reactions have in the main been employed with gas chromatography (GC) to detect trace chemical species or target analytes in complex matrices [15, 16, 17, 18, 19, 20, 21, 22]. Other workers have recently employed "separationless" chemiluminescence methods to determine total sulfur content in gasoline [23] and coal [24] and nitrate and nitrite in flow injection analysis [25]. A very recent supercritical fluid chromatographic (SFC) interface to a chemiluminescent nitrogen detector has also been reported for the examination of polymers and pharmaceuticals [26]; although SFC/chemiluminescence techniques have appeared before [27, 28, 29, 30]. This glancing survey of liquid and gas phase chemiluminescence is not meant to imply that these are the only roles for this method. Researchers have, for instance, recently used in vivochemiluminescence initiated by UV-A irradiation of mouse skin as a means of determining skin oxidative stress processes [31]. Others have used chemiluminescence as a means of following antioxidant evaluation in mouse kidney and brain [32] and plasma [33], as a means of DNA detection and sequencing, detection of polymerase chain reaction-derived nucleic acids, and alkaline phosphatase determination [34]. Finally, chemiluminescence has long been used as a means of measuring concentrations of short lived species in gas mixtures and in the atmosphere and to that end chemiluminescent techniques have been used to determine ozone [35], NOx [36], and hydrogen peroxide [37] in the atmosphere, to detect the possible emitter in the reaction of tetrakis(dimethylamino)ethylene with oxygen [38], and to map out the "hot bands" of HNO produced in the reaction of NO with HCO [39] among many other gas phase applications [40, 41]. The Journal of Bioluminescence and Chemiluminescence (John Wiley & Sons Publisher) is obviously an excellent source in this field and periodically publishes literature searches sorted by year and author [2]. |
Description of a Fluorine-Induced Chemiluminescence DetectorThe experimental results described below were obtained using a gas chromatographic detection system based on the low pressure, gas phase chemiluminescence of the reaction mixture of molecular fluorine with organo-sulfur, -selenium, and -tellurium compounds separated from (gas phase) headspace samples. This detector was originally developed in the research group of John Birks at the University of Colorado, USA [42, 43] and was manufactured and sold by Sievers Instruments (Boulder Colorado, USA). This system can be divided up into three parts: the chromatograph, transfer line, and reaction cell; PMT and photon counting electronics; and the molecular fluorine generator (See Figure 1). |
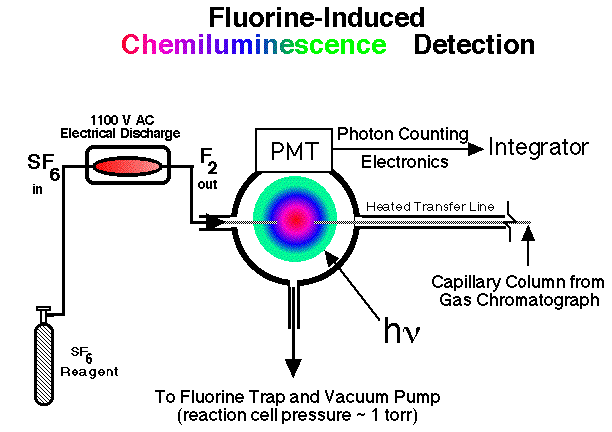 Figure 1. Schematic of the Fluorine-Induced Chemiluminescence Detector |
| Chromatograph and transfer line
A Hewlett Packard 5890 II gas chromatograph (GC) fitted with a 30 m, 0.32 mm i.d. capillary column (DB-5, 0.25 µm film) was used for all chemiluminescence and flame ionization detector (FID) work reported here. The GC injector was maintained at 275°C while the oven was temperature programmed using liquid nitrogen as the oven cryogen. A typical temperature program involved a subambient period to cryogenically trap gaseous samples as a thin band at the head of the column (-20°C) for 1 min, followed by a fast temperature ramp (20°C/min) to a final temperature 25 or 50°C above the highest boiling analyte (usually dimethyl trisulfide, b.p. ~170°C). Instead of being plumbed into our gas chromatograph's normal detector (an FID), the chromatographic column in this system passed through the wall of the GC oven inside a heated transfer line (a 4 mm i.d. nickel tube wrapped with heat tape, thermostatically maintained at ~150°C). The end of the transfer line was attached directly to the cylindrical reaction cell and the column terminated within a mm or so inside that stainless steel cell (reactor volume ~15 cm3). Experimentation has shown that a transfer line temperature greater than 150°C does not improve the chromatographic integrity even for compounds that boil at substantially higher temperatures; however, an increased transfer line temperature does contribute to the warming of the (red sensitive) PMT housing which is adjacent to the reaction cell, and thereby, increases dark current. |
| PMT and photon counting electronics
Figure 2 depicts a schematic of the reaction cell and PMT housing. The PMT used in this work was a red-sensitive Hamamatsu #R2228, end-on photomultiplier tube maintained at a voltage of 1300 V (peak sensitivity ~650 nm, range 300~900 nm). The detector's photon counting electronics have a integration interval, from 2 seconds--adjustable by factors of 2--down to 0.0156 seconds. Signals collected for a chosen period were integrated by a HP 3396 integrator. |
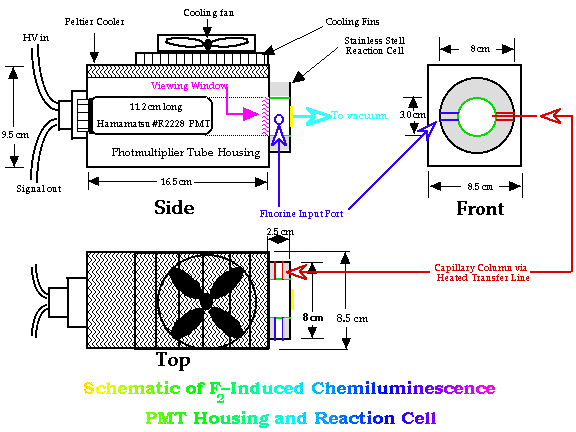
Figure 2. Schematic of Reaction Cell and Photomultiplier Tube Housing of the Fluorine Induced Chemiluminescence Detector |
| Molecular fluorine generator
One of the most unique parts of this system is the method of generating molecular fluorine, F2. Early developmental research with this system by Birks and coworkers involved a microwave discharge of sulfur hexafluoride (SF6) as molecular fluorine source [44], but high backgrounds led the workers to use an alternative source: 5% F2 in He [43]. The inconvenience and safety consideration of maintaining a tank of F2 in an analytical laboratory ultimately lead to another solution: electrical discharge of SF6 (purity 99.996%). Figure 3 shows the physical arrangement of the parts of the small in-line SF6 discharge used in the work reported here. Not depicted in that figure is the flow restricter placed in-line between the sulfur hexafluoride tank and the discharge chamber: at a SF6 delivery pressure of ~15 psi the restricter allows about 1 mL/min SF6 flow into the discharge chamber. |
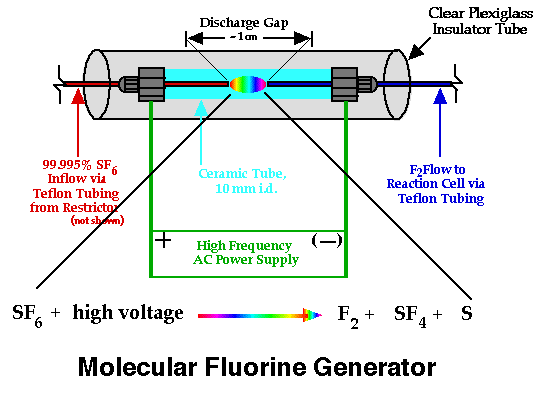 Figure 3. Sulfur Hexafluoride Electrical Discharge |
| Comparison of a detection limit for a standard
organosulfur compound using either 5% F2 in He or the electrical
discharge yields essentially the same detection limit; however, the 5%
F2 in the He system is about 25 to 50% noisier than the discharge
while producing 25 to 50% greater signals for the same mass organosulfur
species; ergo, the same detection limit for both.
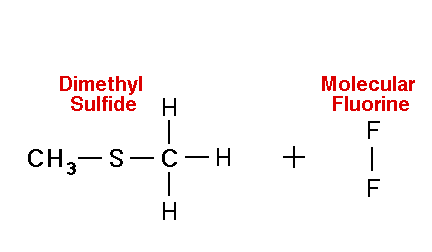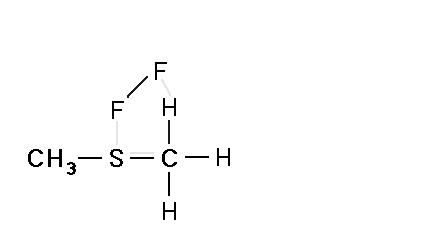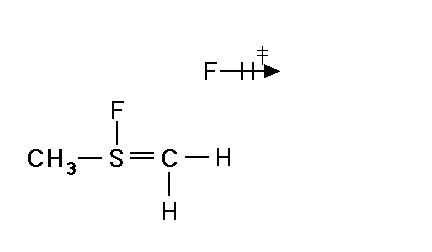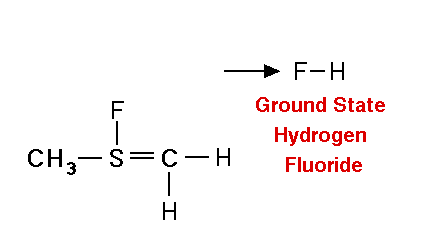
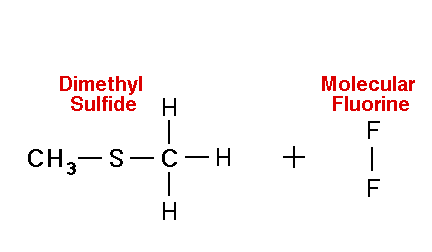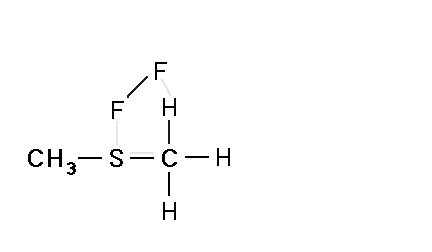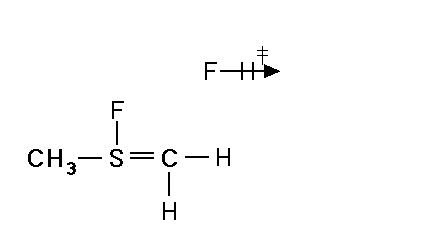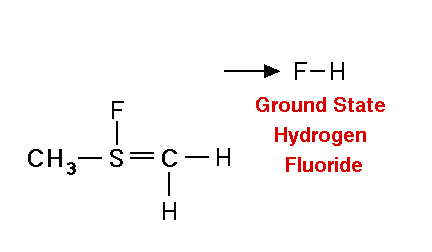
Mechanism for chemiluminescence The mechanism of chemiluminescence light production in the fluorine-induced chemiluminescence detector is not well understood and certainly not as clearly as that for the sulfur/ozone-induced chemiluminescence detector which has been recently studied in detail and in which one major emitter, SO2*, has been detected [20, 17]. This could be because the O3 reaction sequence may be somewhat simpler than the F2-induced chemiluminescent reactions; however, in regards to the O3-induced chemiluminescence system, Burrow and Birks have recently published an intriguing set of experiments which "imply that an unknown sulfur species, X, is formed in the combustion zone and/or transfer line of the instrument and converted to SO upon reaction with ozone in the chemiluminescence detection cell" [45] so maybe things in that system are not as simple as once thought. The hypothesized reaction of molecular fluorine with organosulfur species yields electronically and vibrationally excited HF whose emission signature can be seen in low pressure reaction mixtures of molecular fluorine and methanethiol, CH3SH [46] or ethanethiol, CH3CH2SH [47]. Two mechanisms for the production of HF in this system have been proposed [43, 4, 48]. One possible mechanism can be seen in Figure 4 in which hydrogen fluoride is shown to be the emitting species. This involves a 5-centered reaction intermediate. |
{kind=link}
|
| Probably more important [48]
is a mechanism involving a charge transfer intermediate in which molecular
fluorine accepts an electron from the less electronegative element, sulfur.
The resulting fragmentation process following the formation of the charge
transfer complex subsequently yields various fragments (including excited
HF) which contribute to the chemiluminescence emissions [49,
46, 47]. Therefore
the actual reaction sequence is not simple, and the light actually measured
by a photomultiplier tube focused on a gas phase mixture in this detector
comes from multiple emitting chemical species.
Apparently one of the requirements for the efficient production of detectable light in this reaction is an analyte containing a so-called beta hydrogen which can be abstracted by fluorine as F2 approaches the relatively electropositive heteroatom in detectable analytes (S, Se, Te, etc.). In Figure 4, this b hydrogen is part of the adjacent methyl group. The absence of a b hydrogen in a particular analyte confers little or no detectability for that compound in this detector; therefore, H2S and di-tert-butyl sulfide, for example, exhibit little or no response. Initially applied to organosulfur species [42, 43], this GC detector was later used to determine biogenic organo-selenium and -tellurium [50, 5, 52] and organo-phosphines and phosphinate esters [53]. More recently we have used this system to detect biological organo-antimony production in a monoculture of a Pseudomonassp. and in soil enrichment cultures [54]. |
Materials and MethodsThe following work involves the use of the gas chromatographic/fluorine-induced chemiluminescence detector for the determination of organo-S, -Se and -Te metabolites in the headspace above phototrophic bacteria which were amended with salts and elemental forms of these metalloids. In addition, one of Frederick Challenger's proposed biological intermediate compounds [55], dimethyl selenone (CH3)2SeO2 , was added in parallel experiments to determine if that chemical species would undergo bioreduction to yield dimethyl selenide and dimethyl diselenide.Reagents All reagents were used as received. Sodium selenate (Na2SeO4), sodium selenite (Na2SeO3), and sodium tellurate (Na2TeO4) were obtained from Strem Chemicals (Newburyport, MA, USA). Dimethyl sulfide (DMS), dimethyl disulfide (DMDS), dimethyl selenide (DMSe), and dimethyl diselenide (DMDSe), were purchased from Aldrich Chemical Co. (Milwaukee, WI, USA). Dimethyl telluride was procured from Alfa Products (Danvers, MA, USA). Dimethyl selenone (DMSeO2) was synthesized following the method of Krief et al. [56] with slight modifications that we have been reported elsewhere [57]. The minimal growth medium used (hereafter, Sistrom medium) has also been described elsewhere [58], but briefly, has well-defined buffer and vitamin components and 20 mM succinate as a carbon source at a pH of 6.8 adjusted before autoclaving. Bacterial cultures Six different phototrophic strains (Deutsche Sammlung von Mikroorganismen, Göttingen, Germany) were investigated: Rhodocyclus tenuis (DSM #109), Rhodobacter sphaeroides 2.4.1 (DSM #158), Rhodobacter capsulatus (DMS #1710), Rhodospirillum rubrum S1 (DSM #467) and G9 (DSM #468), and Rhodopseudomonas blastica (DSM #2131). In addition, in one series of experiments the type strain of Rhodobacter sphaeroides 2.4.1 from the American Type Culture Collection (Rockville, MD, USA) was also investigated. Initially made available as stabs (thanks to Professor Reinhard Bachofen, Institute for Plant Biology and Microbiology, University of Zürich, Switzerland) 10 milliliter bacterial cultures were anaerobically grown (Sistrom medium) in 16 mL test tubes sealed with Teflon® coated septa in incandescent light of 10 W/m2 intensity. After inoculation of liquid medium the cultures were left in the dark overnight to deprive them of oxygen before incubation in incandescent light. Ten mg portions of metallic selenium and tellurium were filled into the test tubes prior to autoclaving. These cultures were sampled 7 days after inoculation. The water soluble selenium and tellurium compounds were added from sterile-filtered stock solutions in Sistrom medium. Amendments were added to 10 mL cultures that had grown for two to three days; fresh stock solutions of recrystallized dimethyl selenone were made on the days of amendment [59, 57]. After the amendment these cultures were left in the dark again, and their headspace was analyzed after six to seven days of photoheterotrophic growth. The optical density at 660 nm was used as a measure of biomass. Headspace analyses One mL gas phase samples were removed from the headspace of the test tubes (kept at approximately 25°) by piercing the septum with the needle of a 1 mL gas tight syringe (Alltech Associates, Inc., Deerfield, IL, USA), extracting 1 mL headspace, and immediately injected that gas sample into the hot injector of the GC (275°C). The chromatograph's temperature program was immediately initiated. Chromatography Analysis of volatile, methylated selenium, tellurium and sulfur compounds was carried out by capillary gas chromatography coupled with fluorine-induced chemiluminescence detection using the instrumentation and conditions described above. Retention times of commercial standards were used to identify DMS (CH3SCH3), DMDS (CH3SSCH3) DMSe (CH3SeCH3), DMDSe (CH3SeSeCH3), and DMTe (CH3TeCH3). Flame Ionization Detection A flame ionization detector was used in a few experiments to compare the response of that detector to the response of the fluorine-induced chemiluminescence detector to volatile, biologically produced compounds in bacterial headspace. Cultures of Rhodobacter sphaeroides 2.4.1 were cultured as described above and their headspace gases sampled after 7 days growth. The instrumental conditions (hydrogen, air, and N2 makeup gas settings) of the FID were those specified by the manufacturer. The temperature of the detector was 275°C. The same chromatographic column used in the chemiluminescence work was deinstalled from that detector and reconfigured to the FID for these experiments. The head pressure of the carrier gas (helium) in the FID work was adjusted to yield almost identical retention times for the major organo-sulfur and -selenium analytes (that is, as compared to the retention times in the chemiluminescence chromatography). Different column head pressures are required to achieve similar retention times in chromatography with these two detectors because the FID is basically at atmospheric pressure; while the reaction cell of the chemiluminescence detector where the capillary column ends is approximately 1 torr. |
Results
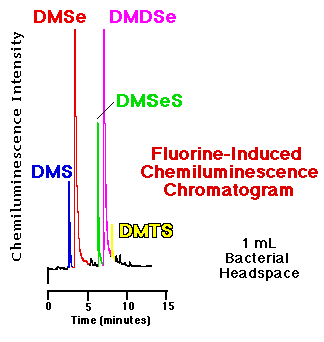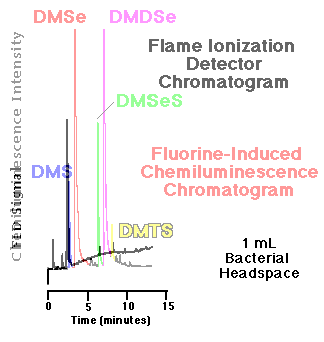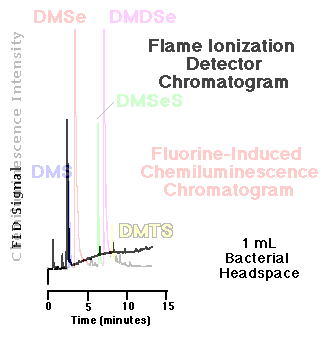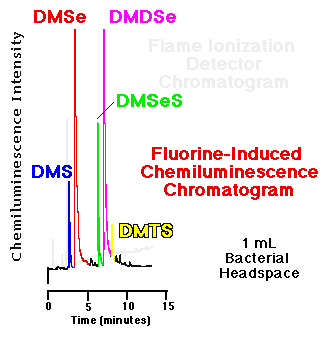
Comparison of an FID and an SCD chromatogram| The response of the flame ionization detector--the most common capillary GC hydrocarbon detector--to organosulfur and organoselenium compounds is substantially less than that of the fluorine-induced chemiluminescence detector. Figure 5 show a comparison of the headspace analyses of replicate, live bacterial cultures amended with 10 mM sodium selenate as determined by each detector. These phototrophic bacteria were grown for seven days before sampling. Peaks routinely quantifiable in the low to middle ppbv range via gas phase chemiluminescence are basically undetectable using FID. | 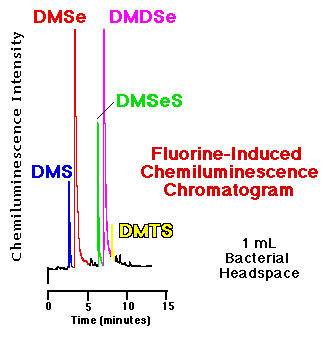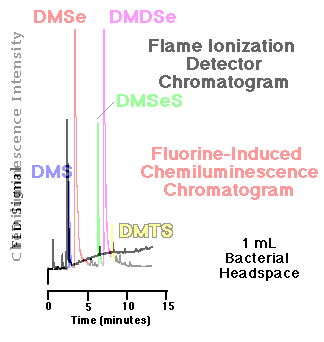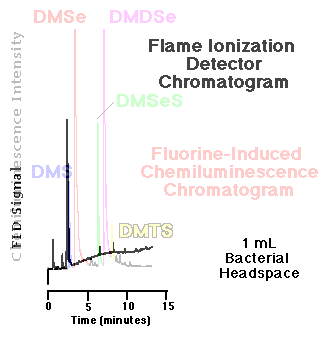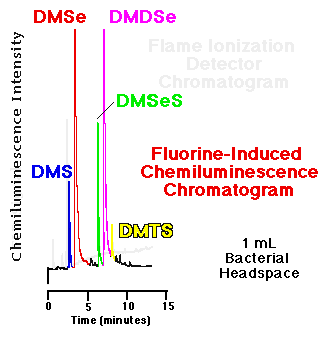
|
{kind=link}
| Selenium amendments
The six bacterial strains investigated were grown for 6-7 days in the presence of 1 mM selenate, 1 mM selenite, or 1 mg/mL metallic Se. Figure 6 displays the results from these selenium experiments. Using optical density as a measure of cell population, some strains were inhibited by the presence of selenium and others enhanced. While all strains produced varying amounts of DMS and DMSe, two of the six strains (R. capsulatus and R. blastica) produced no detectable DMDSe (low parts per billion by volume). Except for cultures of R. tenuis the presence of 1 mM selenate had an inhibitory effect on the production of DMS. |
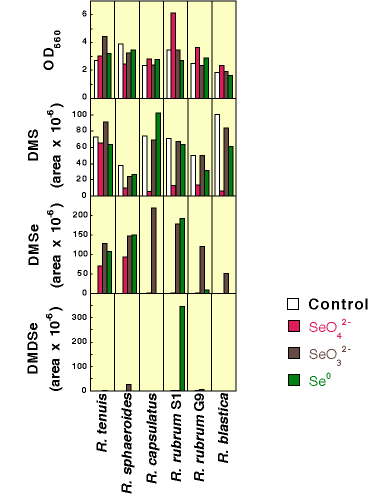 Figure 6. Culture Population and Organosulfur and Organoselenium Production by Six Phototrophic Bacterial Cultures Amended with Selenate, Selenite or Selenium Metal |
{kind=link}
| Dimethyl selenone amendments
As the data in Figure 7 denote, the addition of 1 mM dimethyl selenone to growing cultures of these microbes had no significant effect on growth except in the case of R. blastica and R. sphaeroides. The production of DMS was in all cases positively affected by DMSeO2 amendment at this concentration. Furthermore, as we have seen before [59] organo-selenium production was greatly increased over the organo-Se production by selenate or selenite additions to these same microorganisms in analogous experiments. |
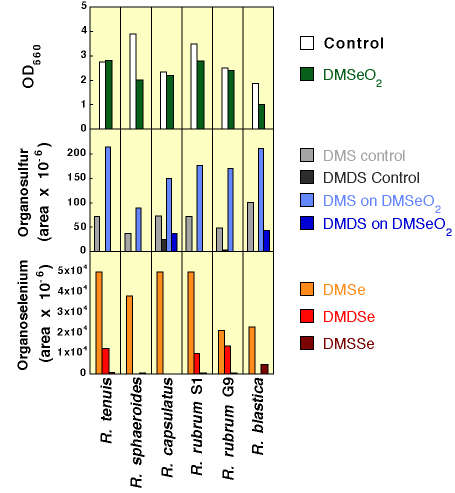 Figure 7. Production of Organosulfur and Organoselenium Compounds by Phototrophic Bacteria Amended with Dimethyl Selenone |
| Tellurium amendments
The biological production of dimethyl telluride was a less common result of adding tellurate to these six microbes (Figure 8). Only R. tenuis, R. rubrum S1, and R. rubrum G9 yielded detectable amounts of DMTe in culture headspace after 7 days incubation. Furthermore, most of this DMTe production came from elemental Te addition (see below). Also of interest is the large amount of DMTe produced by R. tenuis although only selenite yielded headspace DMDSe in the analogous Se experiments. |
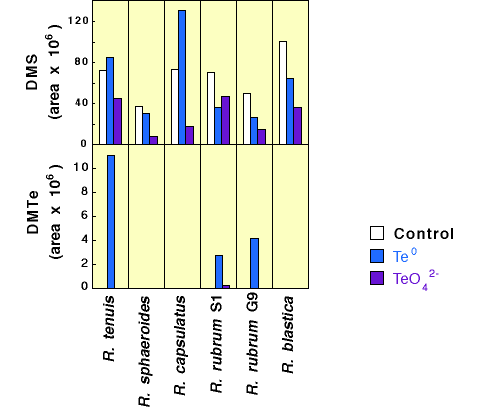 Figure 8. Production of Dimethyl Telluride by Phototrophic Bacteria Amended with Sodium Tellurate and Tellurium Metal |
| Elemental Se and Te
Possibly most surprising, elemental selenium (Se0) and tellurium (Te0) amendments--the additions of insoluble solids which basically remained as insoluble powders at the bottom of the test tube cultures--yielded reduced and methylated Se and Te in four cultures' headspace as Figure 9 shows. Three of these same four strains produced both DMTe and DMSe: R. tenuis and R. rubrum S1 and G9, while R. capsulatus and R. blastica, although growing quite well in the presence of these solids, produced no methylated metalloids in the cultures studied. In the two strains that did not contain detectable amounts of dimethyl selenide or dimethyl telluride, the presence of the elemental metalloids had opposite effects on the production of dimethyl sulfide: In cultures of R. capsulatus more DMS was produced than in the control, whereas in R. blastica less DMS was found than in the control. |
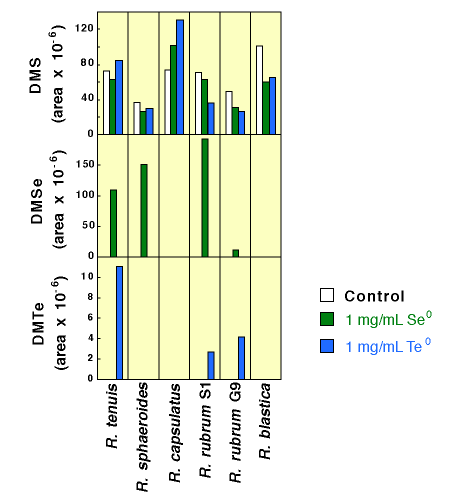 Figure 9. Production of Dimethyl Selenide and Dimethyl Telluride by Phototrophic Bacteria Amended with Selenium and Tellurium Metal |
| Mixed selenate/tellurate amendments
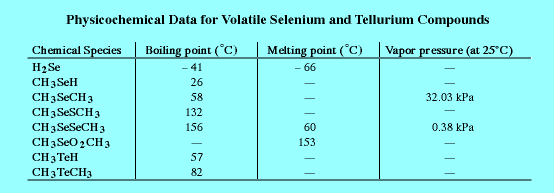
Table 1 shows the summary of experiments involving all strains studied. Also reported here are the first mixed metalloid amendments to microbial cultures of which we know. Expected was the lack of organometalloids in cultures without metalloid salt amendments. DMS was determined in all culture headspace. Cultures amended with sodium tellurate turn black due to elemental Te bioproduction; this even in cultures which did not produce any detectable dimethyl telluride. The presence of 1 mM tellurate in 0.1 mM and 1 mM selenate amended cultures inhibited production of organoselenium dramatically. On the other hand, the combination of 1 mM selenate and 0.1 mM tellurate enhanced the tellurate volatilization (organo-Te production) significantly. DiscussionThe addition of heteroatoms (non-carbon and -hydrogen atoms) to alkanes decreases the response of the flame ionization detector to those heteroatom-containing species. In the case of formaldehyde (CH2O) and carbonyl sulfide (COS) the FID response is basically zero. The reason for this poor FID response is not clearly understood but probably stems from the mechanism of ion formation in the FID flame that involves the formation of CH fragments [60, 61]. All of the analytes reported here contain one or two sulfur or selenium atoms and methyl groups. The differences between these two detectors' response to a mixture of organo-sulfur and -selenium analytes, as seen in Figure 5, is striking and is, in fact, a graphic representation of the reason why specialized detectors are required in GC chromatography.In 1992 Moore and Kaplan reported the high level resistance of proteobacteria to rare-earth oxides and oxyanions [62]. One year later McCarty et al. [63] found dimethyl selenide in the headspace of several strains of purple nonsulfur bacteria, after amending them with sodium selenate. Based on these results we started to investigate six strains of purple nonsulfur bacteria with regard to their capability of methylating and reducing selenium and tellurium compounds. The purple nonsulfur bacteria studied are resistant to metalloid oxyanions at the concentrations investigated here. Cell populations, as measured by optical density, were seldom extensively inhibited. The mutant, Rhodospirillum rubrum G9, however, was an exception. This phototroph is missing a photosynthetic pigment and appears green instead of red like the other strains studied here. The key to this resistance seems to lie in the ability of these organisms to reduce and in some cases methylate the toxic compounds to which they are exposed. Many reduced, methylated metalloids are volatile (boiling points <200°) and in this work were specifically and very sensitively analyzed by fluorine-induced chemiluminescence detection after separation by capillary gas chromatography. Table 2 displays physicochemical data for a few relatively volatile organ-selenium and -tellurium compounds. Compounds with much higher boiling points and small aqueous Henry's Law constants would have vanishingly small presence in culture headspace while still being produced in culture solution. These six strains of purple nonsulfur bacteria were grown under photo-heterotrophic conditions and exposed to varied concentrations of selenite, selenate, and dimethyl selenone, a proposed intermediate of (Challenger's) selenium oxyanion reduction /methylation pathway [55]. After one week the headspace of these cultures was tested for dimethyl selenide, dimethyl diselenide, and dimethyl selenenyl sulfide. In all six cases the highest amounts of volatile selenium compounds were found in cultures amended with dimethyl selenone (Figure 7) and the lowest amounts in cultures doped with selenate (Figure 6). The Challenger mechanism has also been expanded by Reamer and Zoller [64], Doran [65], and Chasteen [66]. Doran's mechanism suggests selenite first be reduced to elemental selenium and then methylated to dimethyl selenide. Thus we also exposed the six bacterial strains to elemental selenium and tellurium to test this hypothesis. In four of the six strains, volatilization was observed upon incubation with elemental selenium (Figure 6). This is a surprising result since the elemental forms of most metals are considered to be biologically stable and relatively inert (Doran's mechanism aside) and therefore to not take part in the cycling of these elements in the biosphere in any significant way. However, storage of elemental sulfur in phototrophic sulfur bacteria (visible as large granules within the cells), e.g. Chromatium okenii, and its possible use as a pool of electron accepting as well as electron donating species has been shown [67]. Our results for Te0 and Se0 suggest that some microbes might be able to reduce and mobilize elemental forms to a greater extent than is presently thought. It has also been put forth that reactions of biologically-produced organoselenium and organosulfur species could exchange and/or disproportionate and thereby produce a mixed organo-S/Se compound, dimethyl selenenyl sulfide (CH3SeSCH3). This compound has been detected in anaerobic and phototrophic headspace [59, 52] and is also reported for 4 of the phototropic strains examined here (Figure 7). Selenium and tellurium belong to group VIB of the periodic table and form very similar compounds with oxygen, hydrogen and methyl groups. Therefore, the bacteria were also amended with tellurate and selenate at the same time in an effort to investigate any synergistic effects of these metalloids. It is known that cultures of purple nonsulfur bacteria amended with tellurium oxyanions turn coal black from elemental tellurium formed by microbial activity [62]. In two of the six strains investigated, dimethyl telluride was found in the headspace after 7 days exposure to sodium tellurate and these two strains also produced dimethyl telluride when they were grown in the presence of elemental tellurium (Figure 8). Furthermore, if selenate and tellurate were added together to cultures of purple nonsulfur bacteria, elemental tellurium was formed; however, in some strains that had not produced DMTe when amended with tellurate alone, mixed SeO42-/TeO42- amendments yielded headspace concentrations of both DMSe and DMTe (Table 1). This suggests to us that the biological response of many of these phototrophs to tellurate to yield DMTe was somehow "turned on" by the co-presence of selenate. Furthermore it appears that the tellurate reduction and methylation to produce DMTe is mainly biological in nature and not simply chemical, that is, caused by the presence of other (reducing) organo-sulfides or -selenides in culture albeit biologically produced themselves. We assert this hypothesis because
ConclusionsFluorine-induced chemiluminescence detection following headspace sampling and separation by capillary gas chromatography is a viable method of determining the effects of toxic metalloid amendment to live bacteria grown in photosynthetic and anaerobic cultures as measured by headspace components. All of the phototrophic bacteria studied here reduced and methylated one or the other oxyanions of Se used and three strains reduced and methylated elemental Te and elemental Se. All strains responded to the addition of dimethyl selenone by producing dimethyl selenide and, in 3 of the 6 strains examined, dimethyl diselenide was also detected. Finally, some of the phototrophic bacteria studied increased their release of dimethyl telluride--produced by their bioreduction and methylation of tellurate--in the presence of selenate. And furthermore the control experiments showed that this synergism was biological and not merely caused by the presence of other organo-sulfides or -selenides in culture.AcknowledgmentsThis research was supported by a Cottrell College Science Award of Research Corporation, the Swiss National Foundation, the Texas Regional Institute for Environmental Studies, Sam Houston State University Research Enhancement Funds, and the Robert A. Welch Foundation. |
{kind=link}