| Photoiupac home page | Discussion | Photobiology.com home |
EFFECT OF SUPERCOLLISIONS ON ENERGY TRANSFER IN
MIXTURES OF BATH GASES AND LASER EXCITED COMPLEX
MOLECULES
Key words: collisional energy transfer, supercollisions, ergodic collision theory of energy transfer, delay luminescence
G. A. Zalesskaya, D. L. Yakovlev, E. G. Sambor, D. I. Baranovsky
Institute of Molecular and Atomic Physics, Academy of Sciences of Belarus,
Scarina av., 70, Minsk 220072, Belarus
ABSTRACT
Intensities and decay rates of delayed luminescence (DL) initiated by a pulse of N2 laser were employed to probe collisional relaxation of complex molecules (benzophenone, acetophenone) diluted with bath gases Ar, Kr, Xe, C2H4, SF6, C5H12. It was shown that vibrational relaxation can be interpreted in terms of two consecutive processes: vibration–vibration (V–V) and vibration–translation (V–T). The results clearly demonstrated that fast component of DL can be used to study V–V energy transfer. It was found that at relatively small internal energy the collisional efficiences of V–V process had the values typical for molecular processes in which supercollisions contribute. The average energies transferred per collision, áDEñ, well correlated with predictions of the simple ergodic collision theory of intermolecular energy transfer.
INTRODUCTION
The determinant role of the intermolecular vibrational energy transfer in chemical kinetic and wide range of physical and photophysical phenomena has long initiated experimental and theoretical studies. Development of quantum electronics, laser chemistry and plasma chemistry stimulated an interest in energy degrading collisions in complicated systems such as vapor of polyatomic molecules. The energy evolution in the real gas phase systems depends on collisional effect, the nature of which has not been clearly understood for complex molecules up to present. Molecular encounters which transfer large quantities of energy in single gas–phase collision, so–called supercollisions, attract particular interest because they can have significant influence in collisional deexcitation of reacting molecules.
The most commonly used methods for observing collisional energy transfer (CET) are time-resolved infrared fluorescence or ultraviolet absorption [1–3]. Electronic excitation followed by fast internal conversion or multiphoton CO2– laser excitation are employed to prepare highly vibrationally excited molecules. In Ref. 4–6 it was shown that time-resolved delayed luminescence could be applied as convenient and sensitive probing of CET providing sufficiently high time resolution. The preparation of vibrationally excited molecule in long–lived triplet state was fulfilled by efficient intersystem crossing S1![]() T1 or direct multiphoton excitation of initially prepared triplet molecules by CO2– laser radiation. The important feature of the present method is that inert bath gases do not influence electronic relaxation processes. In our case vibrational relaxation in singlet manifold was negligible under the bath gas pressure used. Therefore the pressure dependencies of DL decay rates permit to study vibrational energy transfer, if the initial excitation energy is known. But method is restricted to molecules with adequate photophysical properties. The chosen molecules of benzophenone (C13H10O) [7–9] and acetophenone (C8H8O) [10–12] emit DL in gas phase consisting of spectrally overlapping delayed fluorescence and hot phosphorescence with equal decay time. They are characterized by large quantum yields (about unity due to KS @ KTS<KST where KS is a decay rate from S1) and intersystem crossing rates S1
T1 or direct multiphoton excitation of initially prepared triplet molecules by CO2– laser radiation. The important feature of the present method is that inert bath gases do not influence electronic relaxation processes. In our case vibrational relaxation in singlet manifold was negligible under the bath gas pressure used. Therefore the pressure dependencies of DL decay rates permit to study vibrational energy transfer, if the initial excitation energy is known. But method is restricted to molecules with adequate photophysical properties. The chosen molecules of benzophenone (C13H10O) [7–9] and acetophenone (C8H8O) [10–12] emit DL in gas phase consisting of spectrally overlapping delayed fluorescence and hot phosphorescence with equal decay time. They are characterized by large quantum yields (about unity due to KS @ KTS<KST where KS is a decay rate from S1) and intersystem crossing rates S1![]() T1 that have values of about 5×1010s-1 for benzophenone [7] and 1012 s-1 for acetophenone [11]. For small energy gap, such as found in S1
T1 that have values of about 5×1010s-1 for benzophenone [7] and 1012 s-1 for acetophenone [11]. For small energy gap, such as found in S1![]() T1 of benzophenone (ES1=26200 cm-1, ET1=24000 cm-1) and acetophenone (ES1=27500 cm-1, ET1=25750 cm-1), the intermediately dense manifold of mixed states can not act as dissipative quasicontinuum and this provides the reversibility of intersystem crossing. We can use bath gas pressure when vibrational relaxation in singlet manifold is negligible. Then excitation molecules in S1 state with a laser light is a convenient means for preparing vibrationally excited molecules in mixed singlet–triplet states with known amount of vibrational energy Evib= hn–ET, where hn is excitation energy, ET is energy of the triplet state.
T1 of benzophenone (ES1=26200 cm-1, ET1=24000 cm-1) and acetophenone (ES1=27500 cm-1, ET1=25750 cm-1), the intermediately dense manifold of mixed states can not act as dissipative quasicontinuum and this provides the reversibility of intersystem crossing. We can use bath gas pressure when vibrational relaxation in singlet manifold is negligible. Then excitation molecules in S1 state with a laser light is a convenient means for preparing vibrationally excited molecules in mixed singlet–triplet states with known amount of vibrational energy Evib= hn–ET, where hn is excitation energy, ET is energy of the triplet state.
The aim of the present work is to study the influence of the nature of vibrational relaxation processes (V–V or V–T) on energy transfer quantities in mixture of polyatomic molecules (benzophenone, acetophenone) with He, Ar, Xe, C2H4, SF6, C5H12 using time–resolved DL that permits to employ sufficient time-resolution. The most simple and direct method of determining collisional efficiencies b is to measure the pressure dependence of the rate of process under study. The experimental b can then be related to an average energy transfer per collision áDEñ [2,3]. The data obtained are proposed to be used for testing the validity of the statistical theories CET for polyatomic molecules.
EXPERIMENTAL
All experiments were fulfilled on a laser pulse fluorimeter with time-resolution 10-8 sec. Excitation was produced by a pulsed N2 laser with repetition frequency of 10 Hz. The photomultiplier was used as a detector. Emitting light was filtered by an interference filter with a 1.5 nm pass band and by cutoff filters. Signals were recorded by digital oscilloscope connected to IBM PC. Emission decay data acquisition and storage were controlled by a computer. A further signal improvement was obtained by mathematical data processing of about 150 signals.
The substances under studies were stored in the side arm of the 120 mm length, 30 mm dia cylindrical heated quartz cell that was evacuated to less than 10-5 Torr and was kept at constant temperature. The vapor pressure was controlled by the temperature of the thermostated side reservoir.
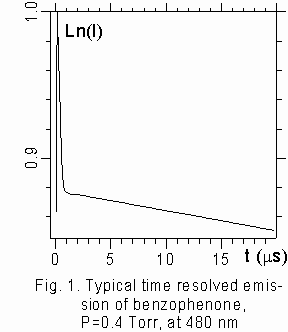 Time-resolved DL of benzophenone and acetophenone at pressures less than 1 Torr exhibited a two-exponential decay behavior characterized by distinct decay times equal to 10 ms and 200 ms for benzophenone, 2 ms and 1200 ms for acetophenone under collisionless conditions (fig. 1). The present data are close to known values [7,8,12]. The adding bath gases causes changes in both the decay rates and time integrated intensities that were estimated as the product of the intensity of the decay curve, extrapolated to t=0, multiplied by corresponding lifetime. Experimental data can be summarized in the following way.
Time-resolved DL of benzophenone and acetophenone at pressures less than 1 Torr exhibited a two-exponential decay behavior characterized by distinct decay times equal to 10 ms and 200 ms for benzophenone, 2 ms and 1200 ms for acetophenone under collisionless conditions (fig. 1). The present data are close to known values [7,8,12]. The adding bath gases causes changes in both the decay rates and time integrated intensities that were estimated as the product of the intensity of the decay curve, extrapolated to t=0, multiplied by corresponding lifetime. Experimental data can be summarized in the following way.
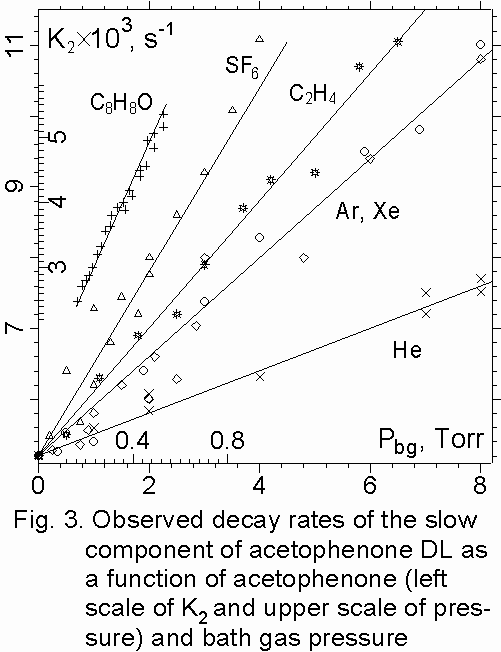
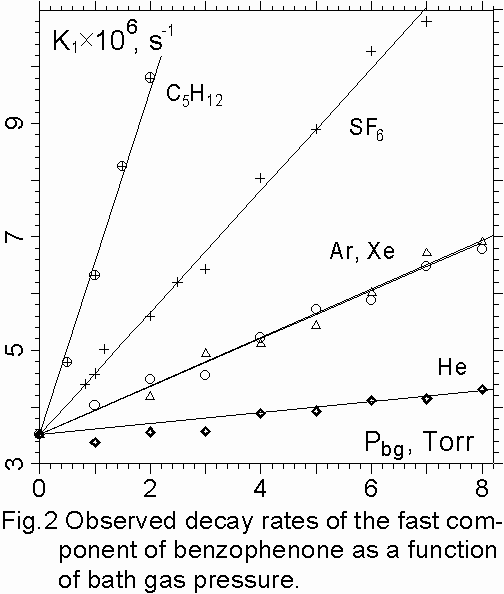 1. DL decay rates of the fast and long components of benzophenone and acetophenone were linearly increasing with vapor pressure over the range of 0.05 – 0.5 Torr for benzophenone and 0.17 – 0.53 Torr for acetophenone and with bath gas pressure over the range of 0 – 8 Torr (fig. 2,3) and following Stern-Volmer behavior. The vapor pressure below 0.05 Torr was not used in the present experiments because the excited molecules can be deactivated on walls.
1. DL decay rates of the fast and long components of benzophenone and acetophenone were linearly increasing with vapor pressure over the range of 0.05 – 0.5 Torr for benzophenone and 0.17 – 0.53 Torr for acetophenone and with bath gas pressure over the range of 0 – 8 Torr (fig. 2,3) and following Stern-Volmer behavior. The vapor pressure below 0.05 Torr was not used in the present experiments because the excited molecules can be deactivated on walls.
 2. The time-integrated intensities of the fast component were decreasing with bath gas pressure satisfying Stern-Volmer relation (fig. 2) I0 /Ibg=1+ Pbg×Z×t, where I0 and Ibg are the time-integrated intensities of the pure vapors and their mixtures with bath gases respectively, Z is the gas kinetic bimolecular collision rate constant, t is lifetime of vibrationally excited molecules. Intensity reductions by bath gases increase as IHe>IAr = IKr > IC2H4 > ISF6 > IC5H12. At bath gas pressure higher than 2 Torr for C5H12 the fast component was disappeared.
2. The time-integrated intensities of the fast component were decreasing with bath gas pressure satisfying Stern-Volmer relation (fig. 2) I0 /Ibg=1+ Pbg×Z×t, where I0 and Ibg are the time-integrated intensities of the pure vapors and their mixtures with bath gases respectively, Z is the gas kinetic bimolecular collision rate constant, t is lifetime of vibrationally excited molecules. Intensity reductions by bath gases increase as IHe>IAr = IKr > IC2H4 > ISF6 > IC5H12. At bath gas pressure higher than 2 Torr for C5H12 the fast component was disappeared.
3. Time integrated intensities of long component do not depend on bath gas pressure over the range of 0–2 Torr in which intensities of fast component were decreasing. The ratio of the integrated intensity in the long time decay to that in the short increased with increasing bath gas pressure. This indicates a collision mechanism can populate the long-lived states.
The collisional efficiency is evaluated as b = Kcol /Z since rate constant satisfies the Stern-Volmer relation. Gas kinetic bimolecular collisional rate constant of Z can be obtained on the assumption of the Lennard–Jones or Stockmayer collision frequency because molecules under study are polar partner (m=2.5 D for benzophenone and m=2.9 D for acetophenone). The parameters of the potentials are unknown and they are estimated by the method recommended in Ref. 13. As a result, we used e /K=554 K, s = 6.5![]() for benzophenone and e /K=554 K, s = 6
for benzophenone and e /K=554 K, s = 6![]() for acetophenone. Note that the value determined by Lennard–Jones potential closely agree with those calculated by Stockmayer potential. Knowing the collisional efficiencies we can also calculate the average energies transferred per collisions áDEñ = b × (E–
for acetophenone. Note that the value determined by Lennard–Jones potential closely agree with those calculated by Stockmayer potential. Knowing the collisional efficiencies we can also calculate the average energies transferred per collisions áDEñ = b × (E–![]() ) =b ×Evib using Evib=5670 cm-1 for benzophenone and 4120 cm-1 for acetophenone. Here
) =b ×Evib using Evib=5670 cm-1 for benzophenone and 4120 cm-1 for acetophenone. Here ![]() is the average energy at the temperature of experiment. As pointed out in Ref. 2,3 this dependence áDEñ on internal energy áEñ of excited polyatomic molecules is true in a case of exponential decay. The characteristics of the collisional processes, the average collision number of Cav sufficient to establish vibrational equilibrium and the Z values obtained are listed in table 1.
is the average energy at the temperature of experiment. As pointed out in Ref. 2,3 this dependence áDEñ on internal energy áEñ of excited polyatomic molecules is true in a case of exponential decay. The characteristics of the collisional processes, the average collision number of Cav sufficient to establish vibrational equilibrium and the Z values obtained are listed in table 1.
DISCUSSION
A detailed discussion of the photophysics of benzophenone and acetophenone has been presented by Bush and al. [9], later Zevenhujzen and Van der Werf [7] and by Berger, Steel [10], Hirata, Lim [11,12] respectively. The intermolecular interaction between the singlet and triplet vibronic levels leads to the formation of a singe manifold of mixed |Sñ-|Tñ states. It is known that both the radiative and nonradiative decay rates of the initially excited states and thermalized triplet states differ. For example, for benzophenone the decay rate Kdec=200 s-1 from the bottom of the "pure" triplet manifold and Kdec=1.8×104 s-1 from mixed states at EST<Evib=3500 cm-1 were reported in Ref. [14] and [7] respectively. It is convenient to divide this manifold into two vibrational energy regions. Triplet manifold includes the pure unmixed triplet vibronic states below EST, singlet – triplet separation energy, and mixed ones above EST, where singlet vibronic inter-
TABLE 1.
Data for Collisions between Exited Molecules and Bath Gases
|
Collider |
D P (Torr) |
Kcol (m s-1× Torr-1) |
Z (m s-1× Torr-1) |
b |
– á D Eñ exp (cm-1) |
– á D Eñ theor (cm-1) |
Cav |
|
|
Benzophenone |
C13H10O |
0.05–0.5 |
9.6 |
26.2 |
0.366 |
2075.2 |
2835 c 2086 d |
3 |
|
C5H12 |
0–2.5 |
2.98 |
22.8 |
0.13 a 0.33 b |
741.1 a 1871.1 b |
2268 c 1047 d |
8 3 |
|
|
SF6 |
0–8.0 |
1.06 |
15.1 |
0.070 a 0.210 b |
398.0 a 1190.7 b |
1113 c 1132 d |
14 5 |
|
|
Xe |
0–8.0 |
0.43 |
14.9 |
0.029 a 0.054 b |
163.61 a 306.2 b |
123 c |
34 18 |
|
|
Ar |
0–8.0 |
0.43 |
15.7 |
0.027 a 0.054 b |
155.31 a 306.2 b |
123 c |
37 18 |
|
|
Acetophenone |
C8H8O |
0.17–0.53 |
7.0 |
18.7 |
0.37 a 0.28 b |
1525 a 1154 b |
1441 c 1439 d |
3 4 |
|
SF6 |
0-1.5 |
3.6 |
13.4 |
0.268 a 0.291 b |
1105 a 1200 b |
518 c 1252 d |
4 3 |
|
|
C2H4 |
0–2.0 |
2.7 |
20.8 |
0.129 |
532 |
285 c 1285 d |
8 |
|
|
Xe |
0–2.0 |
1.6 |
12.4 |
0.129 a 0.192 b |
532 a 793 b |
129 c |
8 5 |
|
|
Ar |
0–2.0 |
1.6 |
14.9 |
0.107 a 0.137 b |
441 a 565 b |
129 c |
9 7 |
|
|
He |
0–2.0 |
0.9 |
25.9 |
0.035 a 0.052 b |
144 a 214 b |
129 c |
29 19 |
a Experimental results are obtained from decay rate dependencies on bath gas pressure.
b Experimental results are obtained from time–integrated intensity dependencies on bath gas pressure.
c Approximation of Ref. 20.
d Approximation of Ref. 21.
acts with number n of triplet vibronic levels. Since in our case the vibronic levels are excited incoherently in the region of the overlapping singlet vibronic levels with small singlet leak (due to above mentioned photophysical properties of molecules under study), the decay DL can be approximately interpreted by considering a simple kinetic description. In such a case the influence ISC S1 ![]() T1 on DL decay is negligible. Therefore we interpret this decay to be composite of change in population due to at least four processes:
T1 on DL decay is negligible. Therefore we interpret this decay to be composite of change in population due to at least four processes:
|
(1) B* (|S>–|T>) |
|
B(|S> - |T>), |
rapid collisional relaxation with rate KV = |
|
(2) B*(|S> – |T>) |
|
B|G>, |
irreversible ISC from initially prepared |
|
(3) B(|S> – |T>) |
|
B|T>, |
relaxation of vibrational distribution to |
|
(4) B(|S> – |T>) |
|
B|G>, |
simultaneously with process (3) irre- |
The system can be fitted to the well known three–level kinetic seheme, if we treat B*(|S> – |T>) and B(|S> – |T>) as single levels:
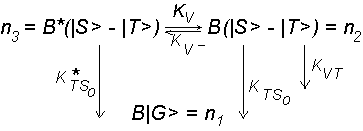
The two necessary differential equations for the above scheme can be written as
![]() (1)
(1)
![]()
The analysis of standart solution to these equations can be restricted only to consideration of the change in population of n3 level, that is given by
![]() (2)
(2)
with the two decay components expressed by Eg. (3); if, as in our case, KV >> KVT,
![]() ,
,
![]() (3)
(3)
It addition, the validity of this assumption was tested by kinetic simulation of the time profiles of DL at different pressure using exact numerical solutions of coupled differential equations. Thus, the fast component of DL has been interpreted both in the earlier works [7–9] and in the present study as coming from a small number of mixed singlet-triplet states initially populated following excitation to levels in S1. The increase of decay rates of the fast component with a pressure growth is determined by the rise of vibrational (V–V) relaxation rates. These conclusions are supported by the pressure dependencies of the integrated intensity of the long decay (I2) and the short one (I1). The results clearly demonstrate that the collisional mechanism can populate the long-lived states. For further check the spectra observed for I1 and I2 have been verified. These spectra have appeared to be similar. It is worth noting that in mixtures of benzophenone with inert gases the collisional efficiency of the fast process is only by one order of magnitude lower than in pure vapors, but it is considerably larger than typical values for V–T relaxation. It is possible that at this stage the collisions with bath gases can only promote nonresonance V–V exchange with vibrational quanta.
Under our experimental conditions, the decay of the slow component is due to V–T energy transfer and ISC from T1 to S0, complications such as triplet- triplet annihilation or photochemical reactions are negligible. It is well known, that the rate KTS0 is a strongly increasing function of temperature which decreases as the bath gas pressure increases. Therefore, increasing Pbg can cause opposite changes of KVT and KTS0. In our case, addition of the low pressures of the bath gases (Pbg < 8 torr) results in increasing the decay rates of the long component, that is the V–T process is rate determined. In such a case the integrated intensity of the long–lived component must also remain constant with growth of Pbg, according to that has been observed for bath molecules. That meant that concentration of the triplet molecules was unchanged. In such a case, the pressure dependence of the long component can be used to estimate the áDEñ values corresponding to V–T CET. For example, the rate constant for V–T process obtained by this way have the values of 2.4×10-2 ms-1×torr-1, 8.3×10-3 ms-1×torr-1 and áDEñ values of 4.4cm-1 and 1.8cm-1 in pure benzophenone and acetophenone; 2.25×10-3 ms-1×torr-1 and áDEñ = 0.8cm-1, 0.3×10-3 ms-1×torr-1 and áDEñ = 0.2cm-1 in mixtures of Kr with benzophenone and acetophenone respectively.
Comparison of these data with the results obtained at CO2 laser multiphoton excitation of triplet molecules has confirmed this interpretation. Essentially, the CO2 laser excitation method to measure the DL quantities has the lower time-resolution and thus it is possible to obtain only the V–T CET quantities as vibrational equilibrium establishes during the CO2 laser pulse. The á D Eñ values for V–T CET obtained by two methods are found to be close. For instance, at CO2 laser excitation of benzophenone mixtures with Kr the á D Eñ values varied from 2 cm-1 to 63 cm-1 with increasing Evib from 3000 cm-1 to 12650 cm-1 [15].
The main experimental results obtained by this technique are summarized as follows: 1) The time–resolution of experiment permits one to divide the intermolecular V–V and V–T relaxation processes for such a large and complex molecules as benzophenone and acetophenone. Two distinguished relaxation time are observed. 2) The ensemble of vibrationally excited molecules becomes vibrationally equilibrium during the fast stage of DL. The results clearly demonstrate that relaxation of upper levels occurs through fast V–V exchange or transfer, which completes after several collisions in mixtures with polyatomic bath gases. 3) At relatively small internal energy (Eint = 9470 cm-1 for benzophenone and 5760 cm-1 for acetophenone), the substantial amount of á D Eñ , typical for supercollisions, is transferred into CET of vibrationally excited molecules with cold one and polyatomic bath gases. 4) In pure vapors and in their mixtures with polyatomic bath gases the collisional efficiencies of V–V process have the values typical for molecular processes in which supercollisions contribute. 5) Based on the V–T data, it was concluded that the majority of energy transfer collisions involve V–T/R transfer of relatively small energies.
Up to now theoretical description of the CET process for polyatomics is lacking. Classical calculations of the requisite quantities have been recently developed to get information on CET [16–18]. In our case, to verify the validity of this, we compared the distribution of internal energy among different vibrational modes according to quantum and classical statistics [19]. It was shown: 1) classical and quantum á Eñ i distributions are different for internal energy used; benzophenone and acetophenone at accessible energy have not reached the classical limit, classical simulation is not valid in this case; 2) high frequency modes of benzophenone and acetophenone are "energy poor", while the low frequency modes are "energy rich".
The experimental data obtained for both molecules demonstrate that after a few collisions with polyatomic gases a vibrational equilibrium was achieved. That is why, the version of the statistical model based on the ergodic assumption [20,21] that after collision the reactant and bath molecule are in microcanonical equilibrium, can be employed to describe the fast V–V CET process. We estimated the á D Eñ since this quantity has been the subject of intensive experimental investigations. The precise predictions by ergodic collision theory [21] require detailed information about the properties of the excited and bath gas molecules. To do this, the temperature dependence of vibrational heat capacities and average vibrational energy of benzophenone and acetophenone were determined preliminarily. The predicted values of á D Eñ presented in table 1 were obtained by equations (51) from Ref. 20 and (31) from Ref. 21. Moreover, the results [21] permitted us to take into account more punctually temperature changes both specific heat and average vibrational energy at a final temperature TC that was determined by the assumption of energy conservation after collisions. As it can be seen (tab. 1), a satisfactory correlation between the energy transfer parameters and the predictions by simple ergodic collision theory were found in the case of V–V energy transfer.
In conclusion, for such large and complex molecules as benzophenone and acetophenone and their mixtures with bath molecules intermolecular V–V and V–T relaxation processes were divided by monitoring time-resolved DL with time resolution 10-8 s. The CET data have been obtained for monatomic and polyatomic bath gases and pure vapors. Attention was called to a large difference in the values of the average energy á D Eñ transferred per collision for V–V and V–T processes. A correlation between the CET parameters and predictions by simple ergodic collision theory was found for the V–V process. At relatively small internal energy, collisional efficiencies of V–V process had the values typical for molecular processes in which supercollisions contribute. It was shown the majority of the energy transfer collisions involve V–T/R transfer of relatively small energies.
REFERENCES
1. Hipler H. and Troe J. in: Bimolecular collisions, ed. J.E. Baggott and M.N.R. AshfoldThe Royal Society of Chemistry, London, 1989.
2. J. Oref, D.C. Tardy, Chem. Rev. 1990; 90: 1407.
3. R.J. Gordon, Comments At. Mol. Phys. 1988; 21: 123.
4. N.A. Borisevich and G.A. Zalesskaya, Spectr. Lett. 1986; 19: 113.
5. G.A. Zalesskaya and A.E. Gololobov, Spectr. Lett. 1993; 26: 1351.
6. G.A. Zalesskaya, D.L. Yakovlev, D.I. Baranovsky and E.G. Sambor, Spectr. Lett. 1996; 29: 1173.
7. D. Zevenhujzen and R. Van der Werf, Chem. Phys. 1977; 26: 279.
8. R.M. Hochstrasser, H. Lutz and G.W. Scott, Chem. Lett. 1974; 24: 162.
9. G.E. Bush, P.M. Rentzepis and J. Jortner, J. Chem. Phys. 1972; 56: 361.
10. M. Berger, C. Steel. J. Amer. Chem. Soc. 1975; 97: 4817–4821.
11. Y. Hirata, E.C. Lim. Chem. Phys. Lett. 1980; 71:167–170.
12. Y. Hirata, E.C. Lim. J. Chem. Phys. 1980; 73: 3804–3809.
13. R.C. Reid, J.M. Prausnitz and T.K. Sherwood, The properties of gases and liquids. McGraw-Hill Book Company, New York, 1977.
14. J.G. Calvert, J.N. Pitts, Photochemistry. Wiley, New-York–London–Sydney, 1966.
15. G.A. Zalesskaya and D.L. Yakovlev, Jurn. Prikl. Spectr. 1995; 62: 1743.
16. R.G. Gilbert, J. Chem. Phys. 1984; 80: 5501.
17. K.F. Lim and R.G. Gilbert, J. Chem. Phys. 1986; 84: 6129, 1990; 92: 1819.
18. G. Lendway and G.S. Shatz, J. Phys. Chem. 1993; 98: 1034.
19. G.A. Zalesskaya, D.L. Yakovlev, D.I. Baranovsky and E.G. Sambor, Spectr. Lett. 1996; 29: 1703.
20. S. Nordholm, B.C. Freiser and D.L. Jolly, Chem. Phys. 1977; 25: 433.
21. L.E.B. Borjesson, S. Nordholm and L.L. Anderson, Chem. Phys. Lett. 1991;186: 65.
Effect of supercollisions on energy transfer in mixtures of bath gases and laser excited complex molecules. G. A. Zalesskaya, D. L. Yakovlev, E. G. Sambor, D. I. Baranovsky (Institute of Molecular and Atomic Physics, Academy of Sciences of Belarus, Scarina av., 70, Minsk 220072, Belarus). Intensities and decay rates of delayed luminescence (DL) initiated by a pulse of N2 laser were employed to probe collisional relaxation of complex molecules (benzophenone, acetophenone) diluted with bath gases Ar, Kr, Xe, C2H4, SF6, C5H12. It was shown that vibrational relaxation can be interpreted in terms of two consecutive processes: vibration–vibration (V–V) and vibration–translation (V–T). The results clearly demonstrated that fast component of DL can be used to study V–V energy transfer. It was found that at relatively small internal energy the collisional efficiences of V–V process had the values typical for molecular processes in which supercollisions contribute. The average energies transferred per collision, áDEñ, well correlated with predictions of the simple ergodic collision theory of intermolecular energy transfer.