AROMATIC AZIDES FOR
PHOTOAFFINITY LABELING:
CRITERION OF THE LONG-WAVE PHOTOSENSITIVITY
Mikhayl F. Budyka and Tatyana S. Zyubina
Institute of problems of Chemical Physics, RAS, 142432 Chernogolovka
(Russia)
E-mail:budyka@icp.ac.ru, zyubin@icp.ac.ru
|
Aromatic azides find a lot of applications in science and technology, and their photochemical properties are widely investigated [1-5]. The majority of aromatic azides absorbs light in UV region of the spectrum. However, for many purposes the azides should be used which are photosensitive in the visible region of the spectrum. Lack of sensitivity to the visible light restricts the application of azides, for example, for photoaffinity labeling in biochemistry and biology, since high energy UV light can destroy biomacromolecules and "mild" visible light should be used instead.
There are two ways to overcome this restriction [6]. The first one consists in application of sensitizers, which absorb visible light and initiate the azide decomposition by intermolecular or intramolecular electron and/or energy transfer [7]. The second way implies synthesis of azidodyes, which long-wave absorption bands lie in the visible region of the spectrum [8]. However, azidodyes have been found to decompose very slowly (quantum yield j ~ 10-5 - 10-3) upon irradiation by visible light.
Considering the aromatic azide sensitivity, the spectral region of the incident light is of principal significance. Irradiation at the long-wave absorption band leads directly to the lowest excited S1 state, and in the case of azidodyes, this state is rather stable relative to the reaction of azido group dissociation. At the same time, azidodyes can undergo effective decomposition upon irradiation by UV light, i.e. when the highest excited states are populated.
At this stage, for the convenience of discussion, we divide aromatic azides into two groups, namely, photoactive and photoinert azides. Photoactive azides decompose upon irradiation at the long-wave absorption band with quantum yields near unity (region 1 > j > 0.1). Photoinert azides are those, which quantum yields of decomposition are less than 0.01 (region 0.01 > j > 0).
We assumed that the electronic structure of photoinert azides should be calculated and compared with that of photoactive azides. The comparison enabled to reveal differences between the two groups of azides and to find the criterion for prediction of the long-wave sensitivity of aromatic azides. Fig. 1 shows the structures of azides studied.
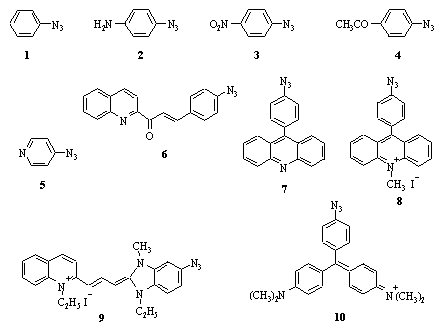
Fig. 1. Structures of azides studied.
The values of quantum yields of azide photodecomposition, both measured by the authors [9-11] and collected from literature, are shown in Table 1.
The structures of azides were calculated with full optimization of geometrical parameters using semi-empirical method MNDO-PM3 [12] (program package MOPAC 7.0). Earlier [13,14], energy parameters for some azides were improved by ab initio RHF, UHF methods with basis 6-31G*; correlation energy was calculated using the second (MP2) and third (MP3) order of Moller-Plesset perturbation theory (GAUSSIAN-94 code was used [15]). Excited states were calculated using CIS and CASSCF levels of theory in GAUSSIAN-94 and EXCITED and C.I. operators in MOPAC 7.0. Details of calculations at ab initio level and comparison with semiempirical method were described in [13,14].
As the first step, using the photoactive azides HN3, PhN3 and 4-NO2C6H4N3 as an example, applicability of PM3 method for description of the process of azido-group dissociation has been shown by comparison of the results of semi-empirical calculations with those of non-empirical ones and with experimental data [13] (there were compared geometrical parameters of azidogroup, the first singlet vertical excitation energies, activation barriers and minimal energy paths of azido group dissociation in the ground and excited states). In all three azides studied, the s-type vacant MO is filled upon excitation to the lowest excited states. This orbital is antibonding on the dissociating N-N2 bond and marked as sNN-one (detail description see in [13]). The filling of sNN-MO gives rise to the characteristic changes in the azido group geometry, significant reduction of the potential barrier of the reaction of molecular nitrogen elimination, and results in the high quantum yield of azide decomposition [13].
As the next step, the set of both photoactive and photoinert azides, from simple substituted phenyl azides to azido-cyanine and azido-triphenylmetane dyes, see (Fig. 1), which long-wave absorption bands covered the region from 250 to 600 nm (Table 1), were considered. The electronic structure of these azides in the ground and lowest excited states has been calculated by PM3 semi-empirical method.
Calculations showed that parameters of the excited states of photoactive and photoinert azides are quite different (see Table 2). On transition from S0 to S1 and T1 states, photoactive azides are characterized by elongation of the N-N2 bond by 0.06 - 0.08 (S1) and 0.13 � (T1), reduction of bond order by 0.25-0.38 (S1) and 0.46-0.48 (T1), decrease of valence angle by 34 - 36 � (S1) and 38 - 42 � (T1), and essential reduction of charge value on the N2 group. On the contrary, in photoinert azides these parameters do not almost change on excitation.
So, in the case of photoactive azides, the structure of the lowest excited states is "prepared" for the dissociation: calculated essential reduction of the N-N2 bond order, and close to zero charge value of the N2 fragment in the excited state of photoactive azides are in accordance with the experimental fact, that during dissociation of azido group, the N-N2 bond cleaves and the charge on the removing N2 molecule is zero.
Analysis of the lowest excited state orbital coefficients revealed that in photoactive azides the sNN-MO is filled. As an example, Fig. 2 reproduces the structure of this orbital in the region of fragment NNN (nitrogen atoms lie in the plane of the paper) and shows the filling of MOs in the S0, S1 and T1 states for the photoactive azide 7 and photoinert azide 8. One can see that in contrast with the photoactive azide, in the photoinert azide the sNN-MO remains vacant.
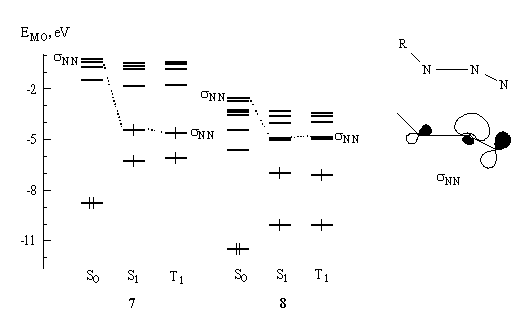
Fig. 2. PM3-calculated structure of the sNN orbital in the region of NNN fragment (nitrogen atoms lie in the plane of the paper) and the filling of MOs in the S0, S1 and T1 states for photoactive azide 7 and photoinert azide 8.
Similar correlation was found for all azides studied ( Table 3): in the photoactive azides sNN-MO is filled, in the photoinert azides the sNN-MO remains vacant (the other type MO is filled) in the lowest excited states (S1 and T1).
Thus, we can connect the experimental data on the photochemical activity of aromatic azides upon excitation to the lowest excited states S1 and T1 with the filling of sNN-MO in this states. This correlation between the ability of aromatic azides to decompose upon irradiation at the long-wave absorption band and the nature of MO filled in the lowest excited states can be used as the criterion for prediction of aromatic azide sensitivity.
In accordance with the correlation found, the criterion can be proposed for prediction of sensitivity of aromatic azides to the visible light. One can expect the azide to be sensitive if its long-wave absorption band is located in the visible region of the spectrum and if sNN-MO is filled in the lowest excited states. Consequently, this azide can be used for photoaffinity labeling by the "mild" visible light. (At this point we should add that photosensitivity is necessary but insufficient condition for photoaffinity labeling: under irradiation azide should also form a reactive intermediate that is able to bind to the substrate. However, nitrene, formed upon azide decomposition, 'trends' sometimes to intramolecular rearrangements rather than to intermolecular labeling).
Table 1. Long-wave absorption maxima (lmax) and quantum yields of azido group decomposition (j) upon irradiation at lexc for azides studied.
Azide |
lmax, nm |
lexc, nm |
j |
Ref. |
1 |
250 |
254 |
0.5 |
10,16-19 |
2 |
- |
- |
0.96 |
20 |
3 |
315 |
313,365 |
0.7, 0.95 |
7,18,19,21 |
4 |
280 |
365 |
0.85 |
7,18,19 |
5 |
270 |
254 |
0.75 |
22 |
6 |
350 |
365,406 |
0.85,0.7 |
9 |
7 |
361 |
365,405 |
0.88,0.69 |
11 |
8 |
430 |
365 |
2.3.10-3 |
11 |
9 |
563 |
563 |
10-5 |
23 |
10 |
620 |
620 |
10-5 |
23,24 |
Table 2. PM3 calculated N-N2 bond length (r) and bond order (p), NNN valence angle and Mulliken effective atom charge for the N2 group (qN2) in the ground and lowest excited states of aryl azides (cations have been calculated without counter ions)
Azide |
State |
r, � |
p |
� NNN, deg. |
qN2 |
1 |
S0 |
1.26 |
1.37 |
169.6 |
0.38 |
S1 |
1.33 |
1.05 |
134.1 |
-0.05 |
|
T1 |
1.40 |
0.91 |
127.5 |
-0.05 |
|
2 |
S0 |
1.26 |
1.38 |
169.6 |
0.38 |
S1 |
1.42 |
0.91 |
152.0 |
-0.06 |
|
T1 |
1.41 |
0.88 |
127.5 |
-0.07 |
|
3 |
S0 |
1.27 |
1.33 |
169.2 |
0.42 |
S1 |
1.35 |
0.98 |
133.5 |
-0.02 |
|
T1 |
1.40 |
0.89 |
127.3 |
-0.02 |
|
4 |
S0 |
1.27 |
1.35 |
169.3 |
0.40 |
S1 |
1.34 |
1.00 |
135.0 |
-0.04 |
|
T1 |
1.40 |
0.88 |
127.5 |
-0.04 |
|
5 |
S0 |
1.27 |
1.35 |
169.4 |
0.40 |
S1 |
1.37 |
0.98 |
134.0 |
-0.03 |
|
T1 |
1.39 |
0.91 |
127.3 |
-0.04 |
|
6 |
S0 |
1.27 |
1.36 |
169.6 |
0.39 |
S1 |
1.33 |
1.11 |
133.3 |
-0.05 |
|
T1 |
1.40 |
0.88 |
127.6 |
-0.05 |
|
7 |
S0 |
1.27 |
1.36 |
169.5 |
0.41 |
S1 |
1.35 |
0.98 |
135.2 |
-0.05 |
|
T1 |
1.40 |
0.90 |
131.1 |
-0.06 |
|
8 |
S0 |
1.28 |
1.29 |
169.7 |
0.45 |
S1 |
1.36 |
1.10 |
168.6 |
0.56 |
|
T1 |
1.27 |
1.34 |
171.0 |
0.43 |
|
9 |
S0 |
1.28 |
1.30 |
170.2 |
0.44 |
S1 |
1.27 |
1.31 |
170.2 |
0.47 |
|
T1 |
1.28 |
1.31 |
170.1 |
0.44 |
|
10a |
S0 |
1.28 |
1.27 |
169.4 |
0.47 |
S1 |
1.28 |
1.31 |
169.9 |
0.43 |
|
T1 |
1.27 |
1.34 |
169.8 |
0.43 |
a model azide with H atoms instead of CH3 groups has been calculated
Table 3. Correlation between the long-wave photochemical activity and filling of the sNN-MO in the lowest excited states of aromatic azides.
Azide |
Activitya |
filling of the sNN-MOb |
1 |
+ |
+ |
2 |
+ |
+ |
3 |
+ |
+ |
4 |
+ |
+ |
5 |
+ |
+ |
6 |
+ |
+ |
7 |
+ |
+ |
8 |
- |
- |
9 |
- |
- |
10 |
- |
- |
a sign “+” denotes that quantum yield of azide photodecomposition j > 0.1, sign “-” denotes that j < 0.01
b sign “+” denotes that sNN-MO is filled, sign “-” denotes that this MO is vacant.
References
1 H.Bayley, J.V.Staros, Photoaffinity labeling and related techniques, in Azides and nitrenes, E.F.V.Scriven, Ed., Academic Press: Orlando, 1984. P.434.
2 D.I.Schuster, et al. Photoaffinity labeling // Photochem. and Photobiol. 1989.- V.49.- N6.- P785-804.
3 J.Brunner, Annu. Rev. Biochem. 1993. V62. P.483.
4 M.S.Platz, Photochem. and Photobiol. 1997.- V.65.- N2.- P.193-194.
5 S.A.Fleming, Tetrahedron, 1995. V.51, P.12479.
6 M.F.Budyka, M.M.Kantor, R.M.Fatkulbayanov, Sci.Appl.Photo., 38, 365 (1997).
7 M.F.Budyka, M.M.Kantor, R.M.Fatkulbayanov, Rus.Chem.Bull., 46, 265 (1997).
8 V.A.Smirnov, S.B.Brichkin, Chem.Phys.Lett., 87, 548 (1982).
9 M.F.Budyka, M.M.Kantor, M.V.Alfimov, Chem.Heterocycl.Comp., 1340 (1991).
10 M.F.Budyka, M.M.Kantor, M.V.Alfimov, Rus.Chem.Bull., 41, 590 (1992).
11 M.F.Budyka, M.M.Kantor, R.M.Fatkulbayanov, Chem.Heterocycl.Comp., 11, 1504-1509 (1997).
12 J.J.P.Stewart, J.Comp.Chem. 1989. V.10. P.221.
13 M.F.Budyka, T.S.Zyubina, J.Mol.Struct. (Theochem), 419, 191 (1997).
14 M.F.Budyka, T.S.Zyubina, M.M.Kantor, Internet J.Chem., 1, Article 2 (1998), http://www.ijc.com/
15 Gaussian 94, Revision D.1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith, G. A. Petersson, J. A. Montgomery, K. Raghavachari, M. A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. B. Foresman, J. Cioslowski, B. B. Stefanov, A. Nanayakkara, M. Challacombe, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defrees, J. Baker, J. P. Stewart, M. Head-Gordon, C. Gonzalez, and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1995.
16 A.Reiser, R.Marley, Trans. Faraday Soc., 1968, V.64, P.1806.
17 A.Marcinek, E.Leyva, D.Whitt, M.S.Platz, J. Am. Chem. Soc., 1993. V.115. P.8609.
18 M.W.Geiger, M.E.Elliot, V.D.Karacostas, T.J.Moricone, J.B.Salmon, V.C.Sideli, M.A.S.Onge, Photochem. Photobiol., 1984. V.40. P.545.
19 A.V.Pochinok, V.Ya.Pochinok, L.F.Avramenko et al., Ukr.Chim.J., 1979. V.45. P.1204 (in Russian).
20 E.A.Pritchina, N.P.Gritsan, N.M.Bazhin, Bull. Acad. Sci.USSR, Chem. Sci., 1986. V.35. P.1587.
21 T.Y.Liang, G.B.Schuster, J. Am. Chem. Soc., 1987. V.109. P.7803.
22 A.V.Eltsov, A.Hanchmann, N.I.Rtischev, J.Org.Chem.(Rus.), 1977, V.13. P.465.
23 V.Ya.Pochinok, V.A.Smirnov, S.B.Brichkin, et al., Ukr.Chim.J., 1984. V.50. P.296 (in Russian).
24 V.A.Smirnov, S.B.Brichkin, M.V.Alfimov et al., High Energy Chemistry, 13, 156 (1979).