Chemiluminescence concomitant with 1,10-phenanthroline-copper/ascorbate/hydrogen peroxide-induced DNA damage
Wenjian Ma En-Hua Cao*
(Institute of Biophysics, Academia Sinica, Beijing 100101,P.R.China)
*Correspondence should be addressed to:
En-Hua Cao
Institute of Biophysics
Academia Sinica
15 Datun Road, Chaoyang District
Beijing 100101, P.R.China
Fax: +86-10-62027837
E-mail: caoeh@sun5.ibp.ac.cn
Key Words: DNA damage; phenthroline-Cu2+ complex ; oxidative stress; reactive oxygen species; chemiluminescence.
Abstract
The chemiluminescence(CL) concomitant with Phen-Cu2+/ascorbate/H2O2-induced
DNA damage was studied. The emission intensity increases linearly with increasing DNA
concentration. An emission spectrum with maximal wavelength at about 410 nm was obtained.
The luminescence was inhibited by histone in a histone concentration dependent manner. The
CL is characteristic of guanine. Of five common bases only guanine can give rise to
luminescence. Investigation of various guanine derivatives show that emission intensity
weakens when N7 and O6 guanine methyl derivatives replace guanine,
whereas it is enhanced when the N9 site is linked with ribose or ribose
phosphate and further enhanced with increasing of phosphate number. Compared with guanine
riboside or guanine nucleotide, guanine deoxyriboside or guanine nucleotide was more
powerful in producing CL. The luminescence was inhibited by all three classes of reactive
oxygen species’ (•OH, ![]() ,
1O2) scavengers with 1O2 scavenger being the
most powerful. By comparing the effect of scavengers on the luminescence of DNA with dGMP,
we propose that •OH might not be the principle species responsible for DNA damage of
above system.
,
1O2) scavengers with 1O2 scavenger being the
most powerful. By comparing the effect of scavengers on the luminescence of DNA with dGMP,
we propose that •OH might not be the principle species responsible for DNA damage of
above system.
Introduction
Free radical-induced DNA damage plays an important role in pathogenesis of human diseases especially in cancer and ageing[1], The copper ion-catalysed reduction of hydrogen peroxide in the presence of a reducing agent, such as ascorbate acid[2,3] or thiols[4], has served as a useful model reaction for generating activated oxygen species, the latter can induce several kinds of DNA damage, including single strand breaks, modified bases, abasic sites, and DNA-protein crosslinks[5]. Strand breakage often occurs near guanine residues, and it has been suggested that Cu2+ ions bind to DNA at these sites[6], based on inhibitor studies[2,3,7], kinetic analysis[8-10], and other means of research[11,12], a site-specific mechanism had been proposed[9], according to this mechanism, copper binding sites on DNA serve as centres for repeated production of reactive oxygen species [ROS], but the nature of the ultimate DNA-oxidising species produced by oxidation of Cu(I)-DNA complexes remains uncertain[10,13,14]. Some researchers have suggested that •OH is the principal reactive intermediate[2-4,15,20], while others disputed this [17,18], and this continues to be a subject of debate[3,10,13,14,19-21].
Most studies to date on the mechanism of DNA damage caused by transition metals have measured breaks[11,12,22-24] or analysed base damage[3,7]. In the present investigation, we have observed a strong chemiluminescence accompanying DNA damaging and the emission is characteristic of guanine. By studying this phenomenon, we hope not only to provide a method of measuring DNA, but to give a new technique in DNA damage research. In addition, as an application, we have investigated the effect of several kinds of radical scavengers and these support the idea that reactive species other than •OH play an important role in DNA damage or, more accurately, in modification of bases. In this paper we report the formation and base specificity of the chemiluminescence concomitant with copper/ascorbate/hydrogen/peroxide-induced DNA damage and the effects of scavengers on the formation of luminescence.
Materials and methods
Reagents. Calf thymus DNA, sodium azide, were purchased from Sigma Chemical Co.; copper sulphate, 1,10-phenanthroline(phen), ascorbate acid, sodium benzoate, sodium formate, hydrogen peroxide were obtained from Beijing Chemical Co.(China); bovine copper-zinc superoxide dismutase (SOD), histone, guanosine and guanine deoxyriboside were obtained from Institute of Biochemistry(Academia Sinica, China); RNA was obtained from British Drug House, Ltd; other bases, nucleosides, nucleotides and polynucleotides were all purchased from Boehringer Mannheim Gmbh; 7-N-methylguanine deoxyribose-5’-phosphate(N7mdGMP) and 6-O-methylguanine deoxribose-5’-phosphate(O6dGMP) were kindly provided by Dr. J.J.Wang.
Chemiluminescence detection. Copper and 1,10-phenanthroline was premixed in 0.1M NaOAc/HOAc(PH5.2) buffer, a moderate concentration of DNA or different bases and nucleotides was incubated with Cu-phen at 18 for 5 minutes. Following this ascorbate and H2O2 were added without interval to the solution to give final volume 1.2 ml. The kinetic curve of chemiluminescence produced in the phen-Cu/H2O2/ascorbate system was immediately recorded with a computerised high-sensitivity single-photon counter(SPC)(type BPCL-4, manufactured in Institute of Biophysics, China.).
Determination of the emission spectrum of the DNA damage. The peak intensities were measured with a fluorescence spectrophotometer with the exciting beam shut down and the kinetic curve of luminescence followed by DNA damage were also recorded with this method.
Determination of the effect of different oxidative radical scavengers and protein. A series of different concentrations of scavengers were premixed in Cu-Phen-DNA solution, after 2 minutes of incubation, the luminescent measurement described above was initiated. Histone and albumin were premixed with DNA for at least 20 minutes before Cu-Phen was added; the other processes were the same as above.
Experiment in D2O as a solvent. 1M NaOAc/HOAc(PH5.2) buffer was diluted by 10 times in D2O solvent, copper and 1,10-phenanthroline was premixed in it, then began the CL measurement as described above. The final concentration were phen: 350M, Cu2+: 50M, H2O2: 160mM, ascorbate: 200M, DNA: 1m g /ml.
Results
DNA Chemiluminescence and its emission spectra
As shown in Fig.1, there is a background chemiluminescence monitored in copper/phenanthroline/ascorbate/H2O2 system(curve 1), which has been assigned to the self-oxidation of phenanthroline by some authors[25], and a delayed emission of light was observed after the addition of DNA into the solution(curve 2), the intensity increased with increasing DNA concentration (shown in Fig.2). Addition of H2O2 alone, Cu2+ alone or ascorbate alone show no chemiluminescence, but when Cu2+-phen and H2O2 are both in solution, DNA Chemiluminescence can be observed, although the intensity is weak. Addition of ascorbate acid to the Cu2+-phen-H2O2 solution, produced a much higher emission intensity ( results not shown), which seems in accordance with the results of Aruoma et al.[3], obtained by analysing the products of DNA damage in this system.
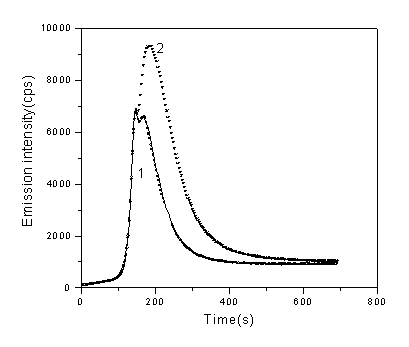
Fig.1 Profile of CL in the absence(curve 1) and presence(curve 2) of DNA (1g/ml), one point every three seconds was recorded which represent the total intensity of this time interval. Concentrations of other regents are CuSO4: 5×10-5 M, Phen: 3.5×10-5 M, ascorbate: 3×10-4 M, H2O2: 160mM, respectively.
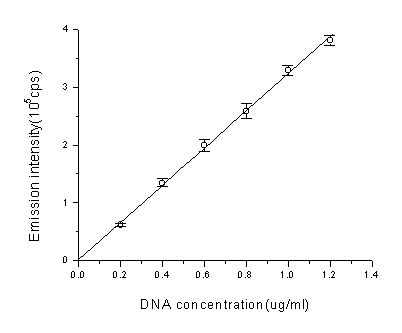
Fig.2 The relationship between emission intensity(total intensity of 900 seconds) and DNA concentration. In each concentration of DNA, the emission intensity was acquired by subtracting the intensity of no DNA from total intensity. Each data is the mean of three independent tests and the bar represents the S.D. Final concentration were phen: 3.5×10-5 M, Cu2+: 5×10-5 M, H2O2: 160mM, ascorbate: 3×10-4 M.
By increasing the DNA concentration, the emission intensity can be made high enough to be monitored by fluorescence spectrophotometry with the excited beam being shut down. As shown in Fig.3, two emission peaks were observed with maximal emissions at 390-410 and 460-480 respectively. The emission spectra of the solution without DNA was also recorded; the maximum emission is around 460nm, which corresponding to the small peak of DNA emission spectra. The build-up and decay of chemiluminescence recorded here on specified wavelength agrees well with the kinetic curve obtained from SPC: two emission peaks were monitored and the light emission at 470nm appeared before the one at 410nm (shown in Fig.4).

Fig.3. The emission spectrum of CL in the absence(a) and presence(b) of DNA. Phen: 3.5×10-5 M, Cu2+: 5×10-5 M, H2O2: 160mM, ascorbate: 3.5×10-4 M, DNA: 20m g /ml.
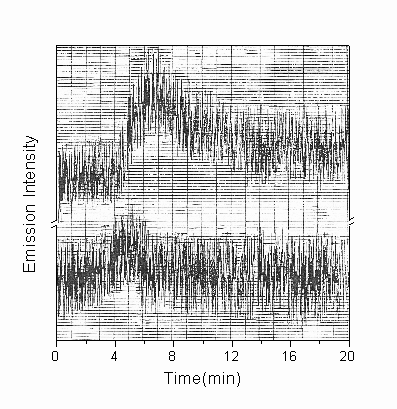
Fig.4. The kinetic curve of CL recorded by fluorescent spectrophotometer in the spectral region of 410nm(a) and 460nm(b), respectively. Experiment conditions were the same as described in the legend to Fig.3.
Effect of histone to the light emission
To confirm the consistence between luminescence and DNA damage, histone was added in solution for its ability to bind DNA specifically. Results showed that the light emission was powerfully inhibited. As a control reagent, albumin showed no effect to the luminescence. Histone or albumin along showed no inhibitory effect on the background luminescence when there is no DNA exist. (shown in fig.5)

Fig.5 Effect of proteins to the CL of DNA.( DNA: 1µg/ml, phen: 3.5×10-5 M, Cu2+: 5×10-5 M, H2O2: 160mM, ascorbate: 3.0×10-4 M). Solid line is the effect of histone, doted line with circle represents the effect of albumen and doted line with cross is the luminescence of background(solution contains no DNA).
The base specificity of chemiluminescence
When DNA was replaced by bases or nucleotides, compared at the same concentrations, of the five common bases(A,G,C,T,U), results showed that only guanine can give rise to Chemiluminescence which was similar to DNA; the same held true to nucleotides (table.1). As shown in Fig.6, there is also a linear relation between emission intensity and guanine concentration.
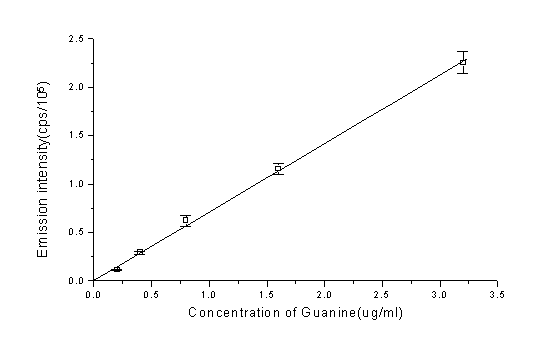
Fig.6 Plot of emission intensity vs. guanine concentration. phen: 3.5×10-5 M, Cu2+: 5×10-5 M, H2O2: 160mM, ascorbate: 3.0×10-4 M. All data were acquired by subtracting the emission intensity (total intensity of 900 seconds) of no guanine from total intensity. Each data is the mean of three independent tests and the bar represents the S.D.
Table.1 The light emission of different bases and nucleotides
Sample |
Emission intensity (104counts) |
Sample |
Emission intensity (104counts) |
A |
0.49± 0.01 |
polyA |
1.23± 0.06 |
T |
-0.36± 0.01 |
polyT |
0.94± 0.02 |
C |
-0.12± 0.006 |
polyC |
0.62± 0.02 |
U |
0.54± 0.01 |
polyU |
1.26± 0.03 |
G |
4.64± 0.2 |
polyG |
10.7± 0.4 |
GR |
14.5± 0.3 |
poly(G-U) |
27.0± 0.8 |
dGR |
17.1± 0.3 |
polyd(G-C) |
45.8± 0.7 |
dCMP |
1.63± 0.08 |
RNA |
12.0± 0.5 |
GMP |
19.8± 0.7 |
DNA |
20.8± 0.9 |
dGMP |
29.4± 0.6 |
O6mdGMP |
1.96± 0.07 |
GTP |
20.6± 0.3 |
N7mdGMP |
1.61± 0.07 |
dGTP |
31.7± 0.5 |
Note: The emission intensity showed here is the total intensity of 900 seconds(the background emission of no nucleotides was subtracted). All values represent the mean ± standard deviation from triplicate measurement.
To understand the mechanism involved in the formation of the Chemiluminescence, the effect of substituting group was determined(shows in table.1). Results were given as follows. (i).Ribose: Comparing the luminescence caused by guanosine(GR), guanine deoxyriboside(dGR), nucleotides and deoxynucleotides (GMP, dGMP, GTP, dGTP), results shows a marked increase of GR or dGR over guanine and the intensity of dGR was higher than GR, as to nucleotides and deoxynucleotides, the latter can give rise to a more powerful of intensity. The overall order is: G<GR<dGR<GMP<GTP<dGMP<dGTP. (ii).Phosphate: the emission intensity of guanine nucleotides is higher than guanosine and intensity increases as phosphate number increases, however, increment rate of intensity becomes lower gradually. The following order is clearly: G<GR<GMP<GTP; G<dGR<dGMP<dGTP. (iii).Methylate: Addition of O6mdGMP or N7mdGMP to the Phen-Cu2+/ascorbate/H2O2 system produce little chemiluminescence, intensity of the two are in equivalence, both are about 15 percent of the dGMP’s intensity in same condition, suggest that the change in molecular structure can make much effect to the luminescence, substituting group in different sites of guanine lead to different results.
The effect of scavengers on the chemiluminescence
As shown in Fig.7, the effect of adding scavengers of oxygen-derived
species upon chemiluminescence which can be a mark of DNA damage was measured. Results
showed that neither •OH scavenger sodium benzoate nor ![]() scavenger SOD had significant effects, while the 1O2
scavenger NaN3 was more powerful, and the same held true when other scavengers
of these three ROS were used (results not shown). In order to verify further whether
•OH is the most important ROS that leads to DNA damage by excluding such effect as
site-specific effect, etc.[3], we used dGMP to replace DNA in this system, to see if there
are any differences when the same scavengers are added. Results showed no significant
changes comparing with the solution of DNA.
scavenger SOD had significant effects, while the 1O2
scavenger NaN3 was more powerful, and the same held true when other scavengers
of these three ROS were used (results not shown). In order to verify further whether
•OH is the most important ROS that leads to DNA damage by excluding such effect as
site-specific effect, etc.[3], we used dGMP to replace DNA in this system, to see if there
are any differences when the same scavengers are added. Results showed no significant
changes comparing with the solution of DNA.
The enhancement of D2O on CL when being used as solvent
To further confirm whether 1O2 is involved in the generation of CL, we use D2O as solvent in this system, results show that the emission intensity was remarkable enhanced to both background and DNA solution and their enhanced level are similar. Compared to the emission intensity (using total intensity of 900 seconds) of CL when using H2O as solvent, the former is about 3 times as high as the latter (shown in Fig.7).
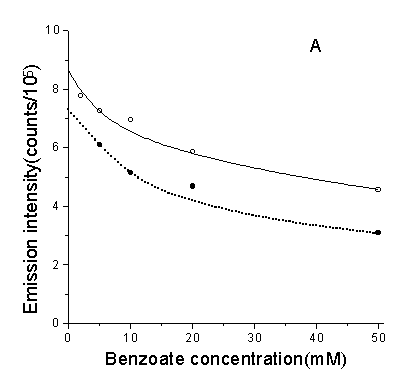
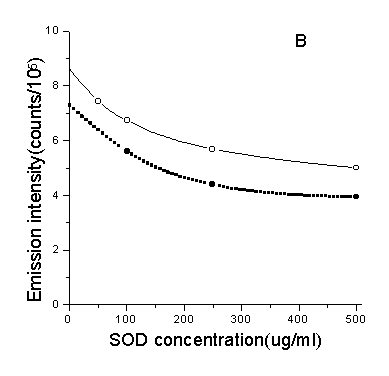

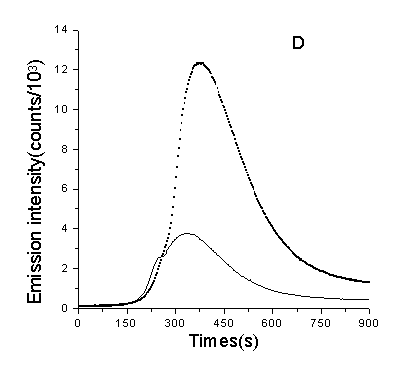
Fig.7 Effect of ROS scavengers and D2O to the CL. A, B, C were the inhibitory effect of three scavengers to the CL of DNA(solid line) (DNA: 1µg/ml, ascorbate: 3.0×10-4 M) and dGMP(doted line) (dGMP: 1µg/ml, ascorbate: 2.5×10-4M) respectively. D was the enhancement of CL when using D2O as solvent. Concentrations of other regents are phen: 3.5×10-5 M, Cu2+: 5×10-5 M, H2O2: 160mM.
Discussion
In this paper, evidence is presented that chemiluminescence appears followed by Phen-Cu/ascorbate/hydroxide-induced DNA damage, and that guanine residues are involved. The consistence between luminescence and DNA damage is definite. First, as shown above, the luminescent species formation is a linear function of DNA or guanine concentration. Second, the luminescence was greatly decreased with addition of histone which binds to DNA in solution, while albumin ( no DNA binding) is utterly useless, and both had no influence to the background luminescence, which is also related to ROS ( shown in Fig.5). In addition, that the maximal emission wavelength of solution containing DNA is shorter than that of background excluded the possibility of energy transfer. All these results imply that the luminescence comes from DNA changes, and that emission intensity can show the level of DNA damage, or at least the level of guanine modification.
As to the mechanism of luminescence, there is little known so far. From
the present work, the following can be concluded. First, the formation of luminescent
species is highly specific since no luminescence at 410 nm appears in the absence of
guanine or when guanine is replaced by other bases or nucleotides. Second, different
substitutes for guanine markedly affect the luminescence. As described above, N9
substitutes, such as ribose, deoxyribose and phosphate, enhanced the luminescence of
guanine, while N7 and O6 methyl derivatives decreased emission
intensity, suggests that guanine modification is the reason of luminescence, and that the
substituted groups most probably influence the formation of luminescent intermediates or
their mode of decay. The guanine specificity seems consistent with the results of Aruoma
et al.[3] who analysed the product of DNA damage; they found that 8-hydroxyguanine is the
major product, although the luminescent intermediate is not known. It seems some
relationship exists between the two substances, more likely the Chemiluminescence results
from the process: intermediate![]() 8-hydroguanine.
Cadet et al.[26] described the generation of 1,2-dioxetanes by 1O2
plus guanosine, which might decompose to chemiluminescent triplet-excited carbonyl
species[27], the emission wavelength of decay of triplet-excited carbonyl is about 420 nm
[28], in our experiment the emission peak of DNA is around 410 nm, they are approximately
the same excluded the possibility of spectra deviation influenced by substitutes. So the
CL species here may also be related to the excited carbonyl species. Moreover, the
effecting of O6, N7 and N9 substitutes to the CL can be
interpreted that they affected either the formation of 1,2-dioxetanes or the formation of
triplet-excited carbonyl species.
8-hydroguanine.
Cadet et al.[26] described the generation of 1,2-dioxetanes by 1O2
plus guanosine, which might decompose to chemiluminescent triplet-excited carbonyl
species[27], the emission wavelength of decay of triplet-excited carbonyl is about 420 nm
[28], in our experiment the emission peak of DNA is around 410 nm, they are approximately
the same excluded the possibility of spectra deviation influenced by substitutes. So the
CL species here may also be related to the excited carbonyl species. Moreover, the
effecting of O6, N7 and N9 substitutes to the CL can be
interpreted that they affected either the formation of 1,2-dioxetanes or the formation of
triplet-excited carbonyl species.
Our experiments ( results not shown) also show that phenanthroline-Cu2+ was important for the formation of CL species. When Cu2+ was replaced by Fe2+, a strong luminescence could be observed in solution containing no DNA, but the CL was quenched when addition of DNA in this system. It seems to indicate that the CL resulted from DNA damage is specifically related to phenanthroline- Cu2+.
Although it is clear that Cu(I)/H2O2 system can
promote biomolecular oxidative damage, the oxidising species produced by reaction of
DNA-Cu(I) complexes with H2O2 remains a controversial issue. On the
basis of "fingerprinting", some authors[3] have proposed that the hydroxyl
radical is the main species responsible for DNA damage, pointing out that no other ROS can
produce such a wide range of modified bases. As to the inability of •OH scavengers in
protecting DNA from damage being observed, they attributed it to site-specific reactions,
which •OH is formed at the sites where Cu(I) binds DNA, and react with DNA rather
than scavengers owing to its high activity. In this study, we checked the above idea by
chemiluminescence methods. Since luminescence can be observed whether in the solution
contains DNA or guanine derivatives and emission intensity indicates the level of DNA
damage, it may be possible to provide some useful information by measuring the ability of
different scavengers in lowering emission intensity in a solution containing dGMP instead
of DNA. because the conditions that lead to site-specific or multi-hit mechanisms do not
exist in this system, and with all molecules being well-distributed, the "
short-distance effect" of •OH is also overcame. If •OH is the main ROS,
then we should observe an increasing ability in lowering luminescence of its scavengers.
However results showed no significant changes being taken place, this seems to indicate
that •OH is not the main damaging species. On the other hand, in our experiment the
emission intensity was significantly decreased when scavengers of 1O2 was
used and markedly enhanced in D2O as solvent. It either suggests that 1O2
is most probably responsible for DNA damage( or more accurate for guanine modification) or
suggests that "a successive-producing mechanism" exists, in which 1O2,
![]() and •OH are produced successively
with 1O2 being the first. Whereas this proposal does not rule out
that •OH is formed in the reaction. As can be seen from the results, •OH
scavengers also show some scavenging ability, although it is much lower than 1O2
or
and •OH are produced successively
with 1O2 being the first. Whereas this proposal does not rule out
that •OH is formed in the reaction. As can be seen from the results, •OH
scavengers also show some scavenging ability, although it is much lower than 1O2
or ![]() scavengers. Moreover, the small
amount of hydroxyl radical produced may be a reason for the fact that a wide range of
modified bases have been observed in the damaging product.
scavengers. Moreover, the small
amount of hydroxyl radical produced may be a reason for the fact that a wide range of
modified bases have been observed in the damaging product.
In summary, we have reported here that Phen-Cu induced DNA damage is accompanied by light emission. Depending on the linear relationship between intensity and DNA concentration, the luminescence can be used as a method in quantitative analysis of DNA, as an investigating technique in DNA damage research, and in the research such as the effect of antioxidants etc. As an application, we argue the ROS species that may responsible for DNA damage in this system. However the exact mechanism of Chemiluminescence is still in the dark, we suggest it be considered in future work and hope it to give much information to the research of transit metal induced DNA damage.
Acknowledgements
We thank the National Natural Science Foundation of China for support of this work.
References:
1.Wallace,S.S. (1994) DNA damage processed by base excision repair. Int. J. Radiat. Biol. 66, 579-589
2.Stoewe, R. & W.A.prutz (1987) Copper-catalysed DNA damage by ascorbate and hydrogen peroxide: kinetics and yield. Free Radical Biol. Med. 3, 91-105
3.Aruoma, O.I., B. Halliwell, E.Gajewski and M.Dizdaroglu (1991) Copper-ion-dependent damage to the bases in DNA in the presence of hydrogen peroxide. Biochem.J.273,601-604.
4.Reed, C.J. & K.T.Douglas (1989) Single-strand cleavage of DNA by Cu(II) and thiols: A powerful chemical DNA-cleaving system. Biochem. Biophys. Res. Commun. 162,1111-1117.
5.Rodriguez, R., R.Drouis, G.P.Holmguist, T.R.O’connor, S.Boiteux, J.Laval, J.H.Doroshow and S.A.Akman (1995) Mapping of copper/hydrogen peroxide-induced DNA damage at nucleotide resolution in human genomic DNA by ligation-mediated polymerase chain reaction. J.Biol.Chem. 270, 17633-17640
6.Sagripanti, J.L. & K.H.Kraemer (1989) Site-specific oxidative at polyguanosines produced by copper plus hydrogen peroxide. J.Biol.Chem. 264, 1729-1734
7.Dizdaroglu, M., G.Rao, B.Halliwell and E.Gajewski (1991) Damage to the DNA bases in mammalian chromatin by hydrogen peroxide in the presence of ferric and cupric ions. Arch. Biochem. Biophys. 285, 317-324
8.Goldstein, S. and G.Czapski (1986) Mechanism and reaction products of the oxidation of copper(I)-phenanthroline by hydrogen peroxide. J. Free radical Biol. med. 2, 3-11
9.Chevion, M. (1988) A site-specific mechanism for free radical induced biological damage: The essential role of redox-active transition metals. Free Radical Biol. Med. 5, 27-37
10.Masarwa, M., H.Cohen, D.Megerstein, D.L.Hickman, A.Bakac and J.H.Espenson (1988) Reactions of low-valent transition-metal complexes with hydrogen peroxide, are the "fenton-like" or not? 1.The case of Cu+aq and Cr2+aq . J. Am. Chem. Soc. 110, 4293-4297
11.John, D.C.A. and K.T.Douglsd (1989) Apparent sequence preference in cleavage of linear B-DNA by the Cu(II): Thiol system. Biochem. Biophys. Res. Commun. 165, 1235-1242
12.Kazakov, S.A., T.G.Astashkina, S.V.Mamaev and V.V.Vlassov (1988) Site-specific cleavage of single-stranded DNAs at unique sites by a copper-dependent redox reaction. Nature. 335, 186-188
13.Yamamoto, K., and S.Kawanish (1989) Hydroxyl free radical is not the main active species in site-specific DNA damage induced by copper(II) ion and hydrogen peroxide. J.Biol.Chem. 264, 15435-15440
14.Milne, L., P.Nicotera, S.Orrenius and M.J.Burkitt (1993) Effects of glutathione and chelating agents on copper-mediated DNA oxidation: Pro-oxidant and antioxidant properties of glutathione. Arch. Biochem. Biophys. 304, 102-109
15.Feldberg, R.S., J.A.Carao & R.Paradise (1985) Recognition of a cytosine base lesion by a human damage-specific DNA binding protein. J. Free Radical Biol. Med. 1, 459-466
16.Rowley, D.A.& B.Halliwell (1983) Superoxide-dependent and ascorbate-dependent formation of hydroxyl radicals in the presence of copper salts: A physiological significant reaction? Arch. Biochem. Biophys. 225, 279-284
17.Sutton, H.C.& C.C.Winterbourn (1989) Formate oxidation induced by a copper peroxo complex produced in fenton-like reactions. Free Radical Biol. Med. 6,53-60
18.Johnson, G.R.A., N.B.Nazhat & R.A.Saadalla-Nazhat (1988) Reaction of the aquacopper(I) ion with hydrogen peroxide: Evidence for a (cupryl) copper(III) intermediate. J. Chem. Soc. Chem. Commun. 407-408
19.Dizdaroglu, M., O.I.Aruoma, B.Halliwell (1990) Modification of bases in DNA by copper ion-1,10-phenanthroline complexes. Biochem. 29, 844 7-8451
20.Czapski, G., S.Goldstein & D.Meyerstein (1988) Free radical Res. Commun. 4, 231-236
21.Aronovitch, J., D.Godinger, A.Samani, G.Czapski (1987) Ascorbate acid oxidation and DNA scission catalyzed by iron and copper chelates. Free Radical Res. Commun. 2, 241-258
22.Tachon, P. (1990) DNA single strand breakage by H2O2 and ferric or cupric ions: Its modulation by histidine. Free Radic.Res.Commun. 9, 39-47
23.Reed, C.J., K.T.Douglas (1991) Chemical cleavage of plasmid DNA by glutathione in the presence of Cu(II) ions. Biochem. J. 275, 601-608
24.Drouin, R., H.Rodriguez, S.W.Gao, Z.Gebreyes, T.R.O’Connor, G.P.Holmquist and S.A.Akman (1996) Cupric ion/ascorbate/hydrogen peroxide-induced DNA damage: DNA-bound copper ion primarily induces base modifications. Free Radical. Biol. Med. 21, 261-273
25.Fedorova, O.S., S.E.Olkin and V.M.Berdnikof (1982) The chemiluminescence mechanism in 1,10-phenanthroline oxidation during catalytic decomposition of hydrogen peroxide. Z. Phys. Chemie. 263(3), 529-549
26.Cadet, J. M.Berger, C.Decarroz, J.R.Wagner, J.E.Vanlier, Y.M.Ginot and P.Vigny (1986) Photosensitized reactions of nucleic acids. Biochimie. 68, 813-834
27.Rohatgi-Mukherjee, K.K (1978) Fundamentals of Photochemistry. (John Wiley & Sons), P192
28.Rongliang Zheng. (1992) Free Radical Biology. (Advanced teaching press, China), P92