Photochemical Properties of
Donor-Acceptor Complexes Formed
by Associated Forms of Porphyrin
A.V. Udal’tsov* and A.A. Churin
Faculty of Biology, Moscow State University, Moscow 119899 Russia
Photochemical properties of meso-tetra(p-aminophenyl)porphine associated in the solution and also absorbance and luminescence properties of the porphyrin in thin films and bound to hydrophobic-hydrophilic copolymers were studied using absorbance, luminescence, EPR, and Raman spectroscopy. The results presented here, together with previous data, show that donor-acceptor complexes are formed between associated molecules of the porphyrin both in thin films and in solution. When porphyrin associated in solution is exposed to visible light, generation of radical ions is observed. The corresponding EPR signals are substantially broadened as compared with those of radical cation and anion of meso-tetraphenylporphine. Under action of blue light, a marked decrease in qauntum yield of the radical ions is observed, whereas intensity of vibrational bands of aminoporphyrin in Raman spectra strongly increases as compared to the bands of solvent. These phenomena are explaned by the competition between dissociation of donor-acceptor complex into radical ions and reception of electron excitation by one of the forms of this complex interacting with water.
Introduction
Numerous studies on different model tetrapyrrole compounds were carried out to understand the molecular mechanisms of operating of these pigments in electron transfer events in the reaction center of photosynthesis [1–7]. Conformational distortions of porphyrin macrocycle associated with substituents at the periphery of the macrocycle and low-energy excited states induced by strongly coupled diporphyrins are of great interest [2–4, 8–9]. Usually, the electron transition in the lowest excited state is observed in the red and near IR spectral regions. The properties of this excited state and the pathways of its deactivation were studied using kinetic spectroscopy [10, 11]. On the other hand, the effect of conformational distortions on the p-electron system of the porphyrin macrocycle was observed for a number of porphyrins [8, 9].
It seems interesting that both similar effects, i.e. disturbing of the p-electron orbitals and development of the low-energy excited state may be observed simultaneously under the formation of donor-acceptor complexes between the associated aminoporphyrin molecules [12]. However, in contrast to diporphyrins with the covalently bound molecules [11, 13] a broad band in the red and near IR region in the corresponding spectra of these aminoporphyrins is observed only under their association in thin films in the presence of water traces [14]. These marked changes in the properties of aminoporphyrin induced by the presence of water traces suggest unusual behavior of porphyrin associates under covalent binding to polymers. Furthermore, a detailed study of photochemical properties of meso-tetra(p-aminophenyl)porphine is necessary for understanding of unusual properties of polymer-bound associates of this aminoporphyrin [15].
In this work, photochemical properties of meso-tetra(p-aminophenyl)porphine associated in the solution and the effect of macromolecular surrounding on donor-acceptor interactions in the porphyrin associate under covalent binding of the porphyrin to hydrophobic-hydrophilic copolymers were studied.
Materials and methods
Synthesis of the compounds shown in the scheme was described elsewhere [16–18]. Dimethylformamide (DMF), dioxane, and other organic solvents were purified by traditional methods [19]. Organic solvents were dried using 4� molecular sieves. Distilled water was used to prepare aqueous-organic solutions. Thin films of associated porphyrins were prepared by solvent evaporation from their solutions until a complete disappearance from CaF2 plate surface was attained.
The UV-visible spectra and differential absorption spectra of porphyrin solutions and thin films were recorded with a Specord M-40 spectrophotometer. Fluorescence spectra were obtained using the equipment described in [20]. Emission measurements were carried out under the same conditions as described in [12]. All emission spectra were calibrated fully for the wavelength response of the equipment.
For the measurements of EPR spectra, solution of porphyrin was placed in quartz ampoule of 3-mm diameter and was illuminated by a 1000-W mercury lamp for 1 min at room temperature. Then the products of the photoreaction were frozen by liquid nitrogen under illumination. For selective illumination standard light filters (USSR) were used. The EPR spectra were recorded with an EPR spectrometer at 77K.
Raman spectra of porphyrins were obtained in DMF solution with a setup described elsewhere [21]. Excitation was provided by a He-Cd laser into the Soret band (lex = 441.6 nm) or by an argon laser in the Qy(1,0) band (lex = 514.5 nm) or in the intermediate band (lex = 488.8 nm). For registration a spectral slit width of 4 cm–1 was used. The measurements were performed at 298 K. Concentration of porphyrin in solutions was lower than 5 � 10–5 M.
For registration of photoinduced changes in the absorption spectra at 298K, solution of porphyrin in 0.2 cm cell was illuminated with a 150-W halogen lamp. The light intensity, Iabs(n), absorbed by a volume unit of the porphyrin solution with wavenumber, n, was calculated as follows:
Iabs(n) = Iirr(n) Tlf(n) (1 – Tsol(n)),
where Iirr(n) is the radiation intensity, Tlf(n) is the transmittance of light filter, Tsol(n) is the transmittance of porphyrin solution. A continuous spectrum of incandescent lamp with the temperature color of 3000K [22] was used for the calculation. Then the total value of light absorbed, Iabs, per an unit of time was calculated. The concentration of radical di-cation was estimated from the difference of absorption after and before illumination at the 500 nm (DA500) using of the extinction coefficient of 0.5 � 105 M–1cm–1. This value was obtained under oxidation of porphyrin II by molecular bromine [23]. The quantum yield of the radical di-cation was calculated at initial stage of illumination from concentration changes with respect to the total value of the absorbed light.
Results
1. Properties of the associated forms of porphyrin in solution, thin film, and after covalent binding to copolymer
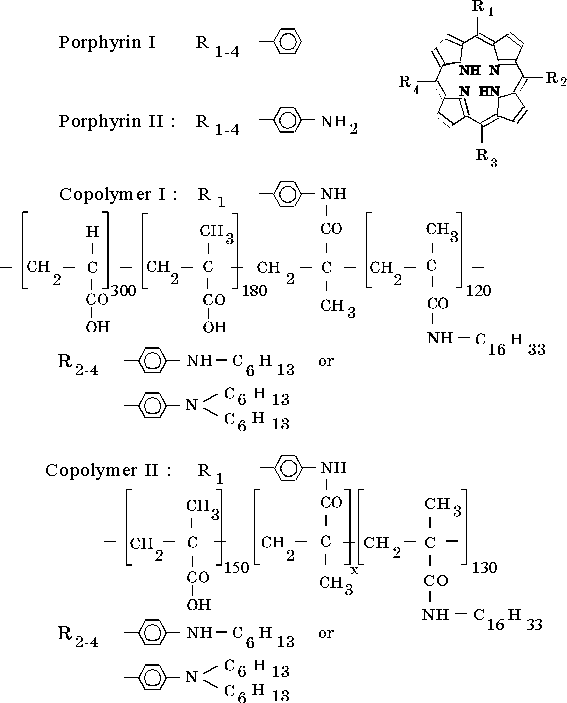
Absorption (Qx electron transition) and fluorescence spectra of porphyrin II associated in DMF are shown in Fig. 1A. The fluorescence spectrum shows the main fluorescence at lmax = 702 nm and two minor emissions, shoulders in the 740 – 800 nm region and in the near infrared region (~ 900 – 1100 nm). The emission in this region reveals donor-acceptor interactions between associated molecules of the porphyrin [12]. Figure 1B shows absorption spectra in thin films (curves 1 and 3) and fluorescence spectrum of porphyrin II in the thin film formed in the presence of water traces (curve 2). As follows from Fig. 1B, the absorption spectra of the film prepared in the presence of water traces (curve 1) and water-free thin film (curve 3) differ each from other. The former shows a new broad absorption band with the maximum at 750 nm. Maximum of the main emission in the fluorescence spectrum of the porphyrin associated in this thin film is observed at 845 nm (curve 2). Furthermore, this spectrum shows the similar shoulder in the near IR region.
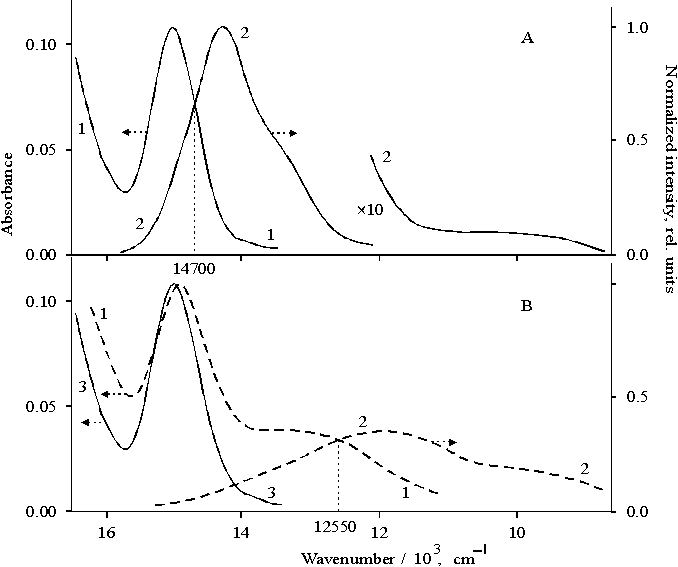
Fig. 1. Absorption (panel A: 1, panel B: 1, 3) and fluorescence spectra (panel A: 2, panel B: 2) of porphyrin II in DMF (panel A: 1 and 2) and in thin films in the presence of water traces (panel B: 1 and 2) and in water-free thin film (panel B: 3).
This evidence shows that different forms of porphyrin associates are produced under different conditions of the development of intermolecular contacts, i.e. in the presence or absence of water traces in thin film. In addition to the main emission related to the deactivation of the excited state via Q electron transition, the fluorescence spectra of the porphyrin solution and thin film show three minor emissions: the emission in the 740–800 nm region (shoulder) (i), in the near IR region, ~ 900–1100 nm (shoulder) (ii), and a broad band with maximum at 845 nm (iii).
Figure 2A presents absorption and fluorescence spectra of porphyrin II bound to copolymer I in DMF, where the degree of association of porphyrin is close to dimeric state [15]. The absorption band of Qx electron transition with the maximum at 652 nm (Fig. 2A curve 1) is narrow as compared with the band in the same transition in the spectrum (Fig. 2B, curve 1) with the maximum at 659 nm where the porphyrin has higher extent of association with respect to the dimer. In the latter case, so called "red tail" is observed. The main emission band of the dimeric form of porphyrin II bound to copolymer I (Fig. 2A, curve 2) is seen at 668 nm, i.e. the band is red shifted by 13 nm as compared with the same band in the spectrum of porphyrin I (curve 3). At the same time, the band of vibronic satellite of this transition observed at 718 nm in the dimer spectrum is not shifted but broadened as compared with the corresponding band in the spectrum of porphyrin I (compare curves 2 and 3). Note that the emission in the near IR region is almost not observed in the spectrum of the dimeric porphyrin bound to copolymer I.
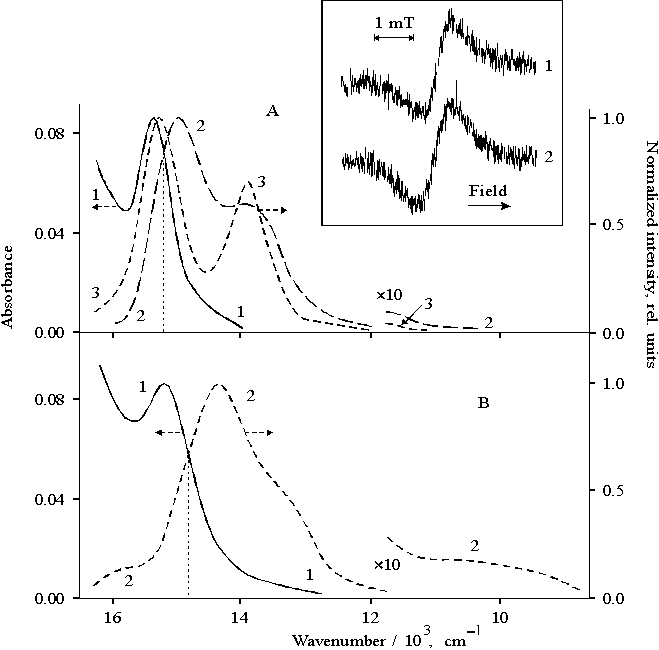
Fig. 2. Absorption (panel A: 1, panel B: 1) and fluorescence spectra (panel A: 2 and 3, panel B: 2) of porphyrin II bound to copolymer I (panel A: 1 and 2) and copolymer II (panel B: 1 and 2); porphyrin I (panel A: 3). The inset shows EPR spectra of porphyrin II bound to copolymer I, 1 and copolymer II, 2 under illumination with visible light (l > 400 nm).
For dimeric form of porphyrin II bound to copolymer I, the EPR spectrum shows the signal with the g-factor of free electron and DHpp = 0.7 mT after visible light illumination (Fig. 2, the inset, curve 1). This EPR signal is related to radical cation of monomeric tetraphenylporphine [24]. In contrast, a broadened EPR signal with DHpp = 1.0 mT appears in the spectrum after the illumination of porphyrin II bound to copolymer II (curve 2) when porphyrin is characterized by a higher extent of association than that of the dimer. In addition to the main band with the maximum at 695 nm, the fluorescence spectrum of porphyrin associate shows the emission at 600–630 nm (a shoulder), the shoulders at 740–800 nm and in the near IR region (Fig. 2B, curve 2). The above minor emissions are absent in the fluorescence spectrum of monomeric porphyrin I (Fig. 2A, curve 3).
Hence, these results indicate that the dimeric and associated states of porphyrin bound to copolymers I and II have their own specific features. The dimeric complex of porphyrin II bound to copolymer I is most likely to dissociate under the illumination with formation of monomer radical ions. Apparently, as a result of the dissociation the near IR emission is practically absent in the fluorescence spectrum. In contrast, the fluorescence spectrum of the associated porphyrin II bound to copolymer II shows the near IR emission, whereas in the corresponding EPR spectrum the abnormally broadened signal is observed as compared to that of monomer radical cation of porphyrin I.
The associated form of porphyrin II bound to copolymer II, which provides the near IR emission, is found to show an unusual behavior under the interaction with quinone. The fluorescence spectra of this sample in the presence of p-benzoquinone are presented in Fig. 3. The main emission with maximum at 695 nm is quenched with increasing the concentration of the quinone whereas the near IR emission appreciably increases (curves 2–4). However, the near IR emission is also quenched at the concentrations of the quinone higher than ~ 60 mM. The plot of the intensity of the main fluorescence band and the near IR emission versus quinone concentration in solution is presented in Fig. 3, the inset. Linear regression analysis of this plot gives the straight line intersecting the y-axis exactly at the unity point (the inset, curve 1). At the same time, the near IR emission is described by the two lines because of the above mentioned features. In the first interval below 60 mM, the experimental values fit the straight line with a negative slope which corresponds to the burning up of the emission. However, extrapolation of this line to the y-axis does not give the intersection at unity. As evidenced by an abrupt slope of the plot (curve 3), an effective quenching of the near IR emission is observed in the second interval.
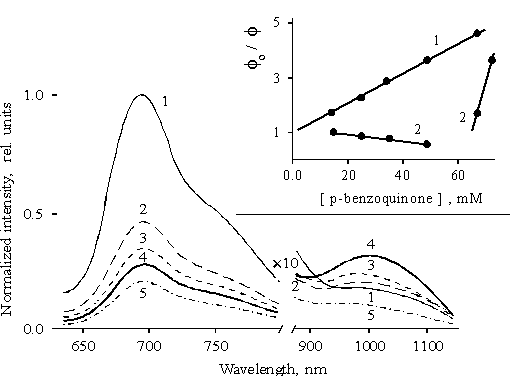
Fig. 3. Fluorescence spectra of porphyrin II bound to copolymer II in the presence of p-benzoquinone in concentrations: 1.5 � 10–2 M, 1; 2.
5 � 10–2 M, 2; 3.5 � 10–2 M, 3; 4.9 � 10–2 M, 4; 6.7 � 10–2 M, 5. The inset shows Stern-Volmer plots for quenching of the main fluorescence band, 1 and the near IR emission, 2, by p-benzoquinone.Hence, the absence of the proportionality between the quenching of the main fluorescence and the burning up of the near IR emission together with an unusual behavior of the near IR emission with changes in the concentration of quinone allow one to suggest that the burning up of the near IR emission is not related to the transfer of excitation from one form of the associated porphyrin to another. An increase in the emission at concentrations of quinone below 60 mM is associated with the quinone involved in donor-acceptor complex with the associated porphyrin, as a result of which the deactivation of the porphyrin increases via the lowest excited state.
2. Photochemical properties of meso-tetra(p-aminophenyl)porphine
Illumination of porphyrin II with the visible light leads to the bleaching of the Soret band in a polar solvent whereas the illumination of the porphyrin in unpolar solvent such as diethylether does not show similar effect. The differential spectrum (the spectrum of porphyrin II in DMF after the 20-min visible light illumination minus the spectrum of the same solution before illumination) is presented in Fig. 4. By its characteristics, this spectrum may be attributed to di-cation of the associated (dimeric) porphyrin II [23] which is accumulated in the solution during illumination. The EPR spectrum of porphyrin II after 1-min illumination recorded at 77K is shown in Fig. 4, the inset, curve 1. In the spectrum, the signal with g-factor of free electron and DHpp = 0.98 mT is overlapped with the broader signal with DHpp = 3 – 4 mT (shown by arrows, curve 1). After defreezing of the solution, both signals disappear in the EPR spectrum (curve 2). The first broadened signal should be associated with radical cation of porphyrin whereas the second broadened signal is likely to be related to radical anion.
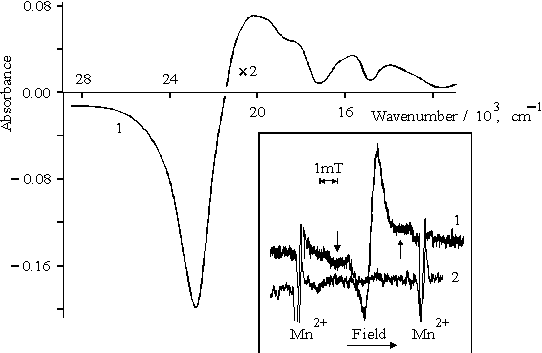
Fig. 4. Differential spectrum of porphyrin II in DMF, 1, see explanation in the text. The inset shows EPR spectra of porphyrin II in DMF under illumination with visible light (l > 400 nm), 1 and after defreezing of the latter, 2.
Hence, these results show that radical-ion pair is formed under the illumination of the porphyrin II with visible light which is likely to dissociate to radical ions in the polar solvent. According to absorption spectra, when illumination exceeds several minutes, the radical cations may associate with the formation and accumulation of di-cation (Fig. 4).
The quantum yield of the formation of the di-cation is found to depend markedly on the spectral composition of light used for illumination (Fig. 5). As compared with the white or filtered light (light filters: B, C, or D), the 3-fold decrease in the quantum yield is observed under violet-blue illumination in the Soret band (light filter: A) (table). Similar ratio between EPR signals is observed in the spectra as a result of violet-blue illumination (Fig. 5, the inset, curve 1) as compared with the visible light illumination (curve 2).
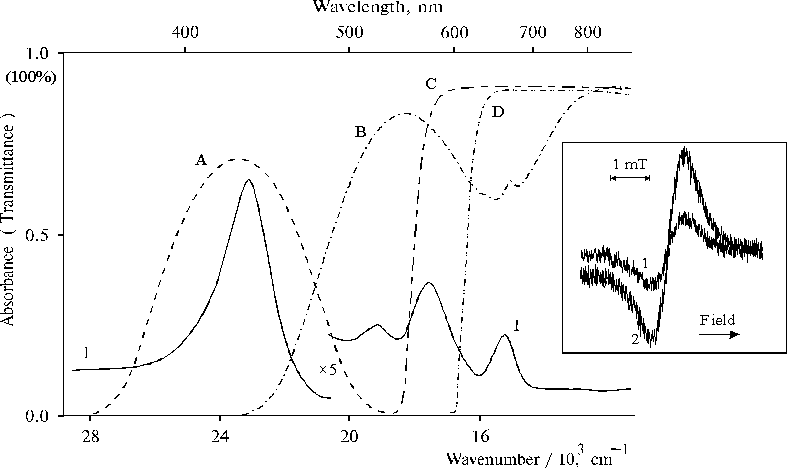
Fig. 5. Absorption spectrum of porphyrin II in DMF, 1 and transmittance spectra of light filters (A, B, C, D) used for light separation. The inset shows EPR spectra of porphyrin II in DMF under illumination with violet-blue light, 1 and visible light (l > 400 nm), 2.
Table
Characteristics of di-cation accumulation at 298K under several minutes illumination of porphyrin II associated in DMF (the porphyrin concentration was 2.2 � 10–5 M).
Light filter |
The integrated intensity of the absorbed light, Iabs |
f di-cation, rel. units |
– |
10 a |
0.9 a |
A |
4.5 |
0.33 |
B |
3.4 |
1.1 |
C |
2.3 |
1.0 |
D |
1.0 |
1.0 |
a – the integrated intensity, Iabs, and the relative quantum yield, f di-cation, were related to those which was calculated under illumination using light filter D.
The effect of the spectral composition of light is also observed in the Raman spectra. Figure 6 shows the Raman spectra of porphyrin II in DMF with lex = 441.6 nm (curve 1) and lex = 514.5 nm (curve 2). The behavior of the Raman spectra of the solvent is independent of the wavelength of excitation (curve 3). A dramatic increase in the intensity of the bands of porphyrin (curve 1) as compared with the intensities of the bands of the porphyrin (curve 2) and the solvent (for example with respect to the band at 868 � 2 cm–1), shows that the blue light excitation is likely to cause strong vibrations in the associated molecules of porphyrin (compare curves 1 and 2). The character of spectrum with the excitation in the region of the Soret band is quite different from that with excitation in the region of quasi-allowed Qy(1,0) transition. Excitation in the intermediate region with lex = 488.8 nm shows the same Raman spectrum as in the latter case.
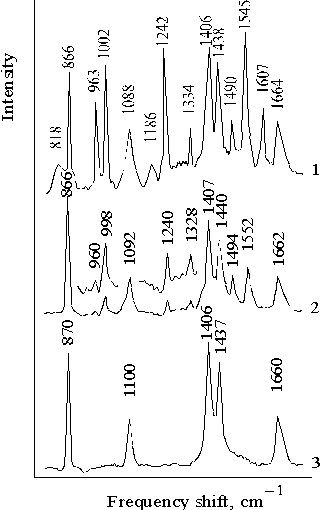
Fig. 6. Resonance Raman spectra of porphyrin II in DMF with excitation at lex = 441.6 nm, 1; lex = 514.5 nm, 2; and dimethylformamide with lex = 441.6 nm, 3. The spectra are normalized on the band of solvent at 868 � 2 cm–1.
These results suggest that the effect of the increased intensity of the vibrational bands in porphyrin macrocycle induced by blue light (lex = 441.6 nm) and a marked decrease in the quantum yield of radical ions under illumination in the Soret band are the result of alternative processes.
3. Interaction of porphyrin dimers with proton
Some properties of porphyrin II in the thin films in the presence of water traces may be simulated by the behavior of porphyrin I without amino groups in phenyl rings in the water-dioxane solutions. The difference between these two porphyrin systems is that during dimerization the molecules of porphyrin I may readily orient with respect to each other whereas orientation of molecules of porphyrin II may be controlled by amino groups because the presence of the amino groups results in strong association of this porphyrin. Furthermore, the results of IR-spectroscopy show that in the thin films of associated porphyrin II, water is involved in the structure of donor-acceptor complex formed by the molecules of this porphyrin [25]. At the same time, in aqueous dioxane solution water may be coordinated by dimer or associate of porphyrin I only in the case of its protonation because phenyl rings without polar groups only enhance the hydrophobicity of the molecule of porphyrin.
In the presence of hydrochloric acid, only protonated forms of porphyrin I are preserved in the aqueous dioxane solution with increasing the content of water. At the same time, neutral molecules of this porphyrin aggregate and precipitate because of its strong hydrophobicity. Figure 7 shows the absorption spectra of dimeric forms of the protonated porphyrin I which was obtained under the competition of the reactions of aggregation and protonation of porphyrin [26]. These spectra show, at least, three forms of the dimers of porphyrin. In the region of the Soret band, these forms have the maxima at 403 nm (violet-form), 465 nm (green-form), and 437 nm. The two latter forms have the maxima in the red region at 696 and 652 nm, respectively. Two of the three forms are singly protonated dimers of porphyrin I (violet and green forms) but the third form with the maximum at 437 nm is doubly protonated dimer. The ratio between these three forms depends on the initial conditions of the aggregation (or dimerization) of porphyrin which competes with the protonation reaction of porphyrin.
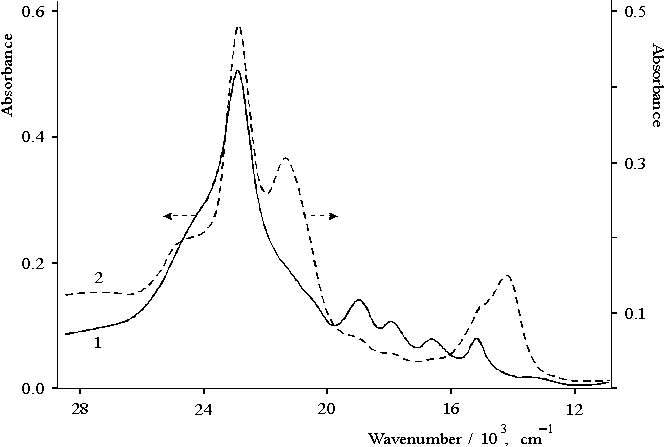
Fig. 7. Absorption spectra of porphyrin I in aqueous dioxane solution in the presence of 0.4 N hydrochloric acid, 1 (1 cm cell) and at higher initial concentration of porphyrin, 2 (0.2 cm cell).
Hence, as a result of protonation reaction competing with the reaction of aggregation of porphyrin in aqueous-dioxane solution there are, at least, three forms of the dimers of porphyrin I interacting with protons, the electron spectra of which considerably differ each from other.
Discussion
According to the fluorescence spectra, the results show that the intermediate forms of the associates of porphyrin II in the excited state are likely to be the consequences of different mode of association of their molecules. Under the selective excitation of porphyrin II in the solution, the corresponding Raman spectra show, at least, two types of associates [12], which differ from each other by the character of intermolecular interactions (Fig. 6, curves 1 and 2). At the same time, the effect of water on the absorbance and luminescence properties of porphyrin II is found to be well-pronounced for thin films of this porphyrin. The presence of water traces in the film markedly changes the electron structure of the associated porphyrin as compared with that of water-free film (Fig. 1). The appearance of a new electron transition with the maximum at 750 nm was attributed to the formation of the donor-acceptor complex, in which proton donating water molecules are involved [25]. The Stokes shift of the emission of this form in the film is found to be very large and equal to 1500 cm–1. Even though this new electron transition has lower-energy, as compared with the quasi-allowed Q-electron transition, no emission with the maximum at ~ 845 nm in the fluorescence spectrum of associated porphyrin II in DMF is observed. Only a high-energy emission in the 740–800 nm region (shoulder) is seen but is still not identified.
Hence, this evidence shows that the radiative deactivation of excitation of porphyrin II associated in thin film in the presence of water traces primarily proceeds via a new low-energy excited state rather than quasi-allowed electron transition. In contrast, the radiative deactivation of porphyrin II associated in the solution proceeds via the Qx electron transition.
The energy of the excited state of the Qx(0,0) electron transition of porphyrin II associated in solution (Fig. 1) is equal to 1.83 eV. The energy gap between Qx(0,0) and 0,0-electron transition of the new low-energy excited state is 2150 cm–1 (or 0.27 eV). The energy of the Qx(0,0) transition in the dimer of this porphyrin bound to copolymer I (Fig. 2A) is found to be somewhat higher than that of associated forms of porphyrin II (Figs. 1 and 2). For dimer and in the associated form of porphyrin II bound to copolymer II the energy is 1.89 eV and 1.84 eV, respectively. The Stokes shift of the emission in the monomer porphyrin I is equal to 240 cm–1 but for the dimer of porphyrin II bound to copolymer I, this value is 370 cm–1 (Fig. 2A). The Stokes shift in associated forms of porphyrin II is two times higher as compared with that in the dimer and is equal to 790 cm–1 for porphyrin II in DMF (Fig. 1A) and to 760 cm–1 for porphyrin bound to copolymer II (Fig. 2B).
Hence, association of porphyrin II leads to a 1.5-fold increase in the Stokes shift for the dimer as compared with that in monomer porphyrin I and a higher (~ 3-fold) increase for the associated forms of porphyrin II. In contrast, the binding of porphyrin II with copolymer II has almost no effect on the Stokes shift as compared with the solution of this porphyrin.
Macromolecular environment of porphyrin bound to hydrophobic-hydrophilic copolymer may assist the formation of intermolecular donor-acceptor complexes. The development of the donor-acceptor complex between p-benzoquinone and porphyrin in the presence of a weak acid in solution is observed only for monomer porphyrin [27]. In the case of dimers or associates of porphyrin, interaction of the both components in the complex should have far more complicated character as compared with monomeric porphyrin, or porphyrin dimer (or associates) may dissociate as a result of the formation of complex with the molecule of quinone. According to the differential excitation spectrum, the radiation center of the near IR emission is a singly protonated associate of porphyrin II bound to copolymer II [12]. Hence, in the associate a strong interaction of porphyrin with carboxyl group of copolymer takes place. Similar character of the fluorescence spectrum in the near IR region (Fig. 3, curve 4) and the fluorescence spectrum of dimeric porphyrin with the maximum at 1000 nm [15] allows one to suggest that the associated (or dimeric) porphyrin interacting with proton also is a radiative center in the presence of quinone in the solution.
Hence, the donor-acceptor complex produced between the associated porphyrin molecules is preserved under the interaction with quinone. Furthermore, as evidenced by an increase of the near IR emission when concentration of quinone is below 60 mM, quinone is involved in the complex with the associated porphyrin. Although, the near IR emission is strongly quenched by the quinone above 60 mM.
The EPR spectrum of porphyrin II associated in DMF suggests the dissociation of intermolecular complex under illumination with visible light to radicals cation and anion (Fig. 4, the inset). Note that violet-blue illumination leads to a marked decrease of the EPR signal (Fig. 5, the inset), i.e the quantum yield of the radicals decreases. These results correlate well with the data for the accumulation of porphyrin II di-cation as revealed by the absorption spectroscopy under steady-state illumination at 298 K (table).
The above results may be explaned as follows. The dimer or associate of porphyrin in the excited state is protonated at one of the pyrrole atoms of the tertiary nitrogen. Such restrictive protonation may lead to intracomplex electron transfer in the excited state. The decay of the intermediate state proceeds via dissociation of the complex to radical ions or by backward electron transfer in the relaxed radical pair with the photon emission in the near IR region. This interpretation is supported by the dissociation of the dimeric complex of porphyrin II bound to copolymer I to monomeric ions under illumination with visible light (Fig. 2, the inset) where DHpp = 0.7 mT exactly corresponds to the monomeric cation radical of tetraphenylporphine [24]. In contrast, the associated forms of porphyrin II, which show the near IR emission, give abnormally broadened DHpp (about 1.4-fold increase) as compared with that in the EPR spectrum of the monomer radical cation.
Hence, a local macromolecular surrounding does not support configuration of porphyrin II dimer when the content of hexadecyl groups is as high as 20% in copolymer I. At the same time, at 40%-content of hexadecyl groups in the composition, a strong association of porphyrin II bound to copolymer II takes place and formation of a stable configuration of the donor-acceptor complex between the associated porphyrin molecules is observed.
The Raman spectra with the selective excitation at lex = 441.6 nm suggest donor-acceptor interaction between porphyrin II molecules associated in solution (Fig. 6). Even though no formation of donor-acceptor complex is seen in solution as is observed in the absorption spectra of thin films (Fig. 1). This fact may be explained by a weak interaction of aminoporphyrin with water in liquid phase as compared with the solid state in the film where rotational and translational mobility is limited. The formation of the configuration of donor-acceptor complex in the solution takes place in the ground state because the configuration of porphyrin molecules in the dimeric complex is produced during association. However, one may hardly exclude that interactions in the associate may acquire donor-acceptor character as a result of illumination. This conclusion is supported by the stage of photoactivation of the initial porphyrin associate during electron transfer to viologen [15], which suggests structural rearrangements of the complex induced by illumination. Furthermore, the formation of the stable restrictively protonated dimeric forms of porphyrin I in the ground state (Fig. 7), when porphyrin macrocycle has not all tertiary nitrogen atoms (=N–) in their protonated state, suggests the existence of a stable configuration, which may serve as a basis for intermolecular donor-acceptor complex.
It seems interesting to compare the intensity of some bands in the Raman spectra at the selective excitation (Fig. 6). The well-pronounced band in the spectrum (curve 1) is observed at 1545 cm–1 and is associated with the planar deformational vibrations of porphyrin macrocycle. The 1552 cm–1 band (curve 2) is also the most pronounced band among the vibrational bands of porphyrin. However, the intensity of this band is lower than those of many bands of the solvent. The 1664 cm–1 and 1662 cm–1 bands (curves 1 and 2) are found to be somewhat broadened as compared with the 1660 cm–1 band in the solvent spectrum (curve 3) where no interaction with porphyrin molecules is observed. In the former case, the 1607 cm–1 band is also observed, and the presence of this band in the IR spectra was explained by the donation of proton by water molecules under the formation of donor-acceptor complex [25]. This activation of deformational vibrations of water suggests that the donor-acceptor interaction between the associated molecules of porphyrin II involves water in the structure of complex with the selective excitation at lex = 441.6 nm. The latter fact is likely to be related to restrictive protonation of the associated porphyrin, i.e., when proton of water or carboxyl group of copolymer is partially or completely transferred to porphyrin [15, 25].
On the other hand, the allowed p-p*-transitions responsible for the Soret band of the restrictively protonated dimeric forms of porphyrin I (Fig. 7) are the result of the electron-vibrational and exciton interactions in the neighboring molecules of dimer, in which the energy of exciton interactions is equal to 1650 cm–1 [26]. Hence, the excitation in the band produced by the allowed electron transitions in dimer (or associate) of porphyrin assists the exciton interaction, the energy of which achieves the energy of deformational vibrations of water. The close values of the energy of deformational vibrations of water molecules and exciton interactions suggest a high probability of the development of resonance interactions with the selective excitation in one of restrictively protonated form of dimeric or associated porphyrin II. In liquid water n2 = 1645 cm–1; in dimethylformamide with minor amounts of water, this value is markedly shifted and equal to about 1660 cm–1 [28]. Hence, when water is involved in the donor-acceptor complex as the proton donor, a new intermediate state activated by light may be produced. In contrast, excitation in the region of quasi-allowed electron transitions does not lead to the similar effect because of weak exciton interactions [29].
Hence, these results show that the formation of radical ions under the blue-light illumination and reception of electron exciation by donor-acceptor complex of the porphyrin molecules, which strongly interact with water to produce a restrictively protonated form, are the competing processes. Therefore, a new intermediate state, which also allows deactivation, is generated under the blue-light excitation; as a result, the quantum yield of radical ions decreases.
Conclusion
The above results show that the donor-acceptor interactions in thin films are stronger where translations and rotations of molecules are limited as compared with those in the solution. In the favorable environment, i.e., in the presence of water traces, a new low-energy excited electron state is formed, the zero-zero transition of which is lower than the corresponding value of the Qx(0,0) electron transition by 2150 cm–1. The dimeric complex of meso-tetra(p-aminophenyl)porphine covalently bound to copolymer I, which contains 20% of hexadecyl groups, dissociates into radical ions under illumination with visible light. In contrast, this porphyrin covalently bound to copolymer II containing about 40% of hexadecyl groups associates in macromolecular surrounding and gives the forms, the configuration of, at least, one of which is supported by the local macromolecular surrounding.
Unusual effects were revealed by studying photochemical properties of meso-tetra(p-aminophenyl)porphine. The formation of radical ions under illumination of the porphyrin solution is observed using EPR spectroscopy. However, the EPR signals are found to be abnormally broadened. According to the EPR spectra, a strong decrease in the quantum yield of the photoinduced formation of radical ions is revealed under the illumination with violet-blue light as compared with that with visible light. This result correlates well with the data on absorption spectroscopy. The considerable change of the Raman spectrum with the selective excitation at lex = 441.6 nm as compared with other excitations (514.5 and 488.8 nm) and a strong decrease in the quantum yield of the radical ions with violet-blue illumination are likely to be related to alternative processes of deactivation. Hence, the reception of the electron excitation by a restrictively protonated form of the donor-acceptor complex formed by porphyrin molecules may compete with dissociation of donor-acceptor complex to radical ions.
Acknowledgement
This work was supported by Russian Foundation of Basic Research, project no. 97-04-48155.
References
1 D.Gust, T.A.Moore, Mimicking photosynthesis, Science 244 (1989) 35–41.
2 O.Bilsel, J.Rodriguez, D.Holten, Picosecond relaxation of strongly coupled porphyrin dimers, J. Phys. Chem. 94 (1990) 3508–3512.
3 A.Osuka, K.Maruyama, N.Mataga, T.Asahi, I.Yamazaki, N.Tamai, Geometry dependence of intramolecular photoinduced electron transfer in synthetic Zinc-Ferric hybrid diporphyrins, J. Amer. Chem. Soc. 112, (1990) 4958–4959.
4 J.-H.Perng, J.K.Duchowski, D.F.Bocian, Effects of steric and electronic interactions on pp overlap in lanthanide porphyrin sandwich complexes, J. Phys. Chem. 94 (1990) 6684–6691.
5 A.M.Brun, A.Harriman, V.Heitz, J.-P.Sauvage, Charge transfer across oblique bisporphyrins: two-center photoactive molecules, J. Amer. Chem. Soc. 113 (1991) 8657–8663.
6 M.R.Wasielewski, Photoinduced electron transfer in supramolecular systems for artificial photosynthesis. Chem. Rev. 92 (1992) 435–461.
7 J. Seth, V. Palaniappan, T.E. Johnson, S. Prathapan, J.S. Lindsey, and D.F. Bocian, Investigation of Electronic Communication in Multi-Porphyrin Light-harvesting Arrays, J. Am. Chem. Soc. 116 (1994) 10578–10592.
8 S. Gentemann, C.J. Medforth, T.P. Forsyth, D.J. Nurco, K.M. Smith, J. Fajer, and D. Holten, Photophysicsl Properties of Conformationally Distorted Metal-Free Porphyrins. Investigation into the Deactivation Mechanisms of the Lowest Excited Singlet State, J. Am. Chem. Soc. 116 (1994) 7363–7368.
9 S. Gentemann, C.J. Medforth, T. Ema, N.Y. Nelson, K.M. Smith, J. Fajer, D. Holten, Unusual picosecond 1(p, p*) deactivation of fuffled nonplanar porphyrins, Chem. Phys. Lett. 245 (1995) 441–447.
10 P.C. Martin, J. Arnold, and D.F. Bocian, Spectroscopic Characterization of Zirconium (IV) and Hafnium (IV) Sandwich Porphyrin Complexes. J. Phys. Chem. 97 (1993) 1332–1338.
11 O.Bilsel, S.N. Milam, G.S. Girolami, K.S. Suslick, and D. Holten, Ultrafast Electronic Deactivation and Vibrational Dynamics of Photoexcited Uranium (IV) Porphyrin Sandwich Complexes, J. Phys. Chem. 97 (1993) 7216–7221.
12 A.V.Udal'tsov, V.Z.Paschenko, A.A.Churin, V.B.Tusov, V.S.Pshezhetskii, Donor-acceptor interactions in porphyrin associates immobilized in biphilic copolymer, J. Photochem. Photobiol. B: Biol., 21 (1993) 87–94.
13 L.L. Wittmer and D. Holten, Photophysics of Lanthanide Triple Decker Porphyrin Sandwich Complexes, J. Phys. Chem. 100 (1996) 860–868.
14 A.V.Udal'tsov, L.A. Kazarin, Stabilization of Donor-Acceptor Complexes Formed by Associated Porphyrins in Thin Films, J. Photochem. and Photobiol. A: Chem. 96 (1996) 99–107.
15 A.V.Udal'tsov, Characteristics of donor-acceptor complexes formed in porphyrin-polymer systems and their photoactivation in electron transfer photoreaction. J. Photochem. and Photobiol. B: Biol. 37 (1997) 31–39.
16 A.D. Adler, F.R. Longo, J.D. Finarelli, J. Goldmacher, J. Assour, and L. Korsakoff, A simplified synthesis for meso-tetraphenylporphin, J. Org. Chem. 32 (1967) 476.
17 J-H. Fuhrhop, and K.M. Smith, Laboratory methods. In K.M. Smith (ed.), Porphyrins and Metalloporphyrins, Elsevier, Amsterdam, 1975, pp. 757–869.
18 A. V. Udal'tsov, Modeling of primary events of photosynthesis in porphyrin-polymer systems, PhD Thesis, Moscow State University, Moscow 1990 pp.84–94 (in Russian).
19 A. Gordon, and R. Ford, Sputnik Khimika, Mir, Moscow, 1976, p.437 (in Russian).
20 A.A. Krasnovsky (Jr.), Photoluminescence of singlet oxygen in pigment solutions, Photochem. Photobiol. 29 (1979) 29–36.
21 Z. I. Gadgiev, A. A. Churin, V. Z. Paschenko, and L. B. Rubin, Raman spectrograph for biological investigations, Biol. Nauki, 8 (1980) 98–104 (in Russian).
22 J. Rabek, Experimental methods in photochemistry and photophysics, Mir, Moscow, 1985, v. 1, pp 40–58 (in Russian).
23 A.V. Udal'tsov, V.S. Pshezhetskii, Photoinduced formation of dimer cation radicals of porphyrin, Khim. Fizica 7 (1988) 1656–1660 (in Russian).
24 R.H. Felton, Redox Reactions of Metalloporphyrins, In: The Porphyrins, D. Dolphin, Ed. Academic Press, New York, 1979, Pt. C, v. 5, pp 53–125.
25 A.V.Udal'tsov and L.A. Kazarin, Influence of water on spectral properties of porphyrin associates in thin films, Biochemistry (Moscow) 61 (1996) 367–373
26 A.V.Udal'tsov, Absorbance and Luminescence Spectroscopy of Restrictively Protonated Dimeric Forms of Porphyrins, Biochemistry (Moscow), accepted.
27 T. Ueda, A. Tanaka, M. Igarashi, and H. Harada, Photo-generated stable complex with efficient power of photo-induced charge separation between porphyrin and quinone, Spectrochim. Acta, 42A (1986) 209–214.
28 J.R. Scherer, Adv. Infrared Raman Spectrosc. 5 (1978) 149.
29 G.P. Gurinovich, E.I. Zenkevich, A.M. Shulga, E.I. Sagun, A. Suisalu, Deactivation of electron excitation in chemical porphyrin dimers, Zh. prikladnoi spectrosc. 41 (1984) 446–455 (in Russian).