The partial molar volume of the proton in water determined by laser-induced optoacoustic studies.
Claudio D. Borsarelli and Silvia E. Braslavsky*
Max-Planck-Institut für Strahlenchemie, Postfach 10 13 65, D-45413 Mülheim and der Ruhr, Germany
Keywords: proton-transfer; proton partial molar volume; photoacoustics; time-resolved volume changes
*To whom correspondence should be addressed
Abstract
The structural volume change, DVMLCT, associated with the formation and decay of the metal-to-ligand charge transfer (MLCT) state of (2,2'-bipyridine)tetracyanoruthenate(II) complex, Ru(bpy)(CN)42-, was studied as a function of pH in dilute aqueous solutions ( < 0.4 M) using time-resolved laser-induced optoacoustic spectroscopy. The values of DVMLCT for the MLCT formation of the complex strongly depend on pH, ranging from +3.0 ± 0.3 cm3/mol at pH = 0.35 to +15.0 ± 0.5 cm3/mol at pH = 6.63. The reduction of DVMLCT at low pH values is assigned to the fast excited-state proton ejection undergone by monoprotonated Ru(bpy)(CN)42- . With the DVMLCT values and the Drude-Nernst equation for the electrostriction effect with an empirically determined constant, a value for the "absolute" partial molar volume of the hydrated proton in water of V°H+ = (-5.5 ± 0.8) cm3/mol is obtained, in good agreement with literature data.
Introduction
In order to determine partial molar volumes it is generally necessary to measure with great precision the density, r, or the density difference between the solution and the pure solvent which in general results in large uncertainties, especially at high dilution. Other methods use the pressure dependence of the ionic equilibria using high-pressure techniques.1-3 However, pressure-dependent methods are not convenient for the study of fast reactions, such as photoinduced electron and proton transfer reactions, since many parameters affecting the reaction change by the increase in pressure, e.g., the solvent compressibility and the structure of the excited state.1-2
These problems are overcome by using time-resolved laser-induced optoacoustic spectroscopy (LIOAS) for the study of enthalpy and reaction volume changes associated with photochemical reactions at normal pressure. LIOAS is a highly sensitive method with time resolution ranging from a few nanoseconds to microseconds.4,5 The experiment consists of measuring the pressure wave evolving in a medium after pulsed laser excitation. The pressure wave contains a thermal term due to radiationless deactivation processes and dependent on the thermoelastic parameters of the solution, and a term due to the possible structural volume changes which is independent of these parameters.4,6
Recently, we have used LIOAS for the determination of the structural volume changes taking place during the quenching of the metal-to-ligand charge transfer (MLCT) state of Ru(bpy)32+ by Fe(III) in aqueous solutions in the presence of various salts.7 The structural volume changes have been interpreted on the basis of a chemical model which takes into account the speciation parameters of the redox reaction in each case. The partial molar volumes at high dilution of several transient species could thus be determined. Such properties are not attainable by any other technique.
Few LIOAS studies on photoinduced proton transfer reactions have been reported so far. The structural volume changes reported by Kurian and Small8 upon excitation of pyranine (8-hydroxypyrene-1,3,6-trisulphonate) in water and in various buffer solutions at pH = 7 were dependent on the nature of the buffer solution, which was attributed to varying interactions between the ejected proton and the buffer molecules and/or the water molecules and the pyranine anion.8 Recently, Bonetti et al.9 reported a volume of 24.5 cm3/mol for the reaction producing water after proton photoejection from o-nitrobenzaldehyde in basic solutions. We are not aware of other time-resolved measurements of partial molar volumes upon proton photoejection.
By convention, the standard partial molar volume of the proton in water is V°H+,conv = 0 at all temperatures. All the absolute values for ions of charge z are referred to this value by a linear equation adding the conventional partial volume to z times the "absolute" partial molar volume of the proton, V°H+. The determination of this "absolute" value therefore becomes extremely important. In a recent review several methods for the determination of ionic volumes in solution have been discussed,10 and special consideration was devoted to the value of V°H+ and the difficulties associated with its determination.
Proton ejection is of obvious biological interest, since it bears similitude with proton translocation through biological membranes in processes such as ATP synthesis. Indeed, in the case of bacteriorhodopsin, photoexcitation of the pigment leads to translocation of the proton needed for this synthesis.11
In the present work we report the application of LIOAS for the calculation of V°H+ in aqueous solutions. To this aim, the structural volume changes (DVMLCT) associated with the formation and decay of the MLCT state of (2,2'-bipyridine) tetracyanoruthenate (II) complex, Ru(bpy)(CN)42-, were monitored as a function of the proton concentration in water. Ru(bpy)(CN)42- was a suitable choice for our study since the photophysical properties of the complex strongly depend on the pH.12
Materials and Methods
The potassium salt of (2,2'-bipyridine)tetracyanoruthenate (II), Ru(bpy)(CN)42-, was kindly supplied by Professor Franco Scandola (Ferrara). The dye bromocresol purple (BCP, Fluka) was used as a calorimetric reference in HCl aqueous solution due to the similarity of its spectrum at pH < 4 to that of the sample. All solutions were freshly prepared before the measurements, and the pH was adjusted (0.3 < pH < 7) with concentrated HCl (Merck). Water was deionized. The pH value was measured for each solution before and after the LIOAS experiment and no variation was observed in any case. The concentration of Ru(bpy)(CN)42- was ca. 5´10-5 M. The absorbances of reference and sample solutions were recorded with a Shimadzu UV-2102PC spectrophotometer and matched within 3% at 400 nm. The solutions of Ru(bpy)(CN)42- were deoxygenated by bubbling for 15-20 min with solvent-saturated argon.
Steady-state emission spectra were recorded with a Spex Fluorolog spectrofluorometer in deoxygenated solutions.
The temperature range for the LIOAS experiments was (15-35) ± 0.1 °C. The LIOAS set-up has been already described.13 In short, the excitation source was a 15-ns (FWHM) laser pulse at 400 nm. A 40-mm polyvinylidene fluoride film was used as the acoustic detector. The signals were amplified 100 times and fed into a transient recorder. The laser beam width was shaped with a 0.2 mm slit so that the effective acoustic transit time was ca. 120 ns which allowed for a time resolution of 10-15 ns using deconvolution techniques. In all cases, linear dependence with zero intercept of the LIOAS signal amplitude with the laser fluence was observed in the energy range studied (up to 40 mJ/pulse). In order to avoid multiphotonic processes, the LIOAS measurements were performed at laser fluences between 10 and 20 mJ/pulse. No photobleaching of the sample and the reference solutions was detected after each experiment.
The procedures used for the LIOAS signal analysis have been described in several publications.4,6,13-15 Often, the photoacoustic signal contains contributions from several kinetic processes occurring within the time resolution of the LIOAS experiments. In these cases, the acoustic signal S(t) is the convolution between the instrument response function, R(t), and a time-dependent pressure decay function P(t), which is assumed to be a sum of single exponential functions. A deconvolution procedure is required in order to recover the amplitude factors, ři , and the lifetimes, ti, for each ith decay step in the sample. In order to perform the deconvolution, a calorimetric reference is used, which converts all the absorbed energy into prompt heat.4,13-15 Within this framework the ři value, normalized to the amplitude of the reference compound, is related to the thermoelastic parameters of the media by eq. 1,
El ři = qi + FR,i DVR,i (cpr/b) (1)
where El is the absorbed photon energy (299.3 kJ/mol at 400 nm), cp the heat capacity at constant pressure, r the mass density, b the cubic expansion coefficient, FR,i the quantum yield, and DVR,i the structural volume change per mole for the ith decay step. The first term on the right side, qi, is related to the enthalpy change during the reaction step i, and can be separated from the second term by varying the ratio (cpr/b). In aqueous solutions advantage is taken of the fact that b exhibits a very strong temperature variation between 3.9 °C and room temperature.
Signal analysis was performed using the program Sound Analysis 3000 version 1.13 (Quantum Northwest), and the amplitude and lifetime values were obtained by averaging from two series of measurements at three different fluence values for each solution. This procedure yields errors lower than 10% in the values derived from the LIOAS experiments.
Since aqueous solutions containing added electrolytes have thermoelastic parameters different from neat water, the correction of the (cpr/b) ratio of the solutions at different temperatures was performed by measuring the LIOAS signals for the calorimetric reference in neat water and in the solution used. Both signal amplitudes are related by a known simple equation.16 The LIOAS signal from the BCP solution at pH 4 was identical to that of an aqueous solution of matched absorbance of another calorimetric reference, K2Cr2O7 , at the same temperature. Thus, the value of the ratio of thermoelastic parameters, (cpr/b), in aqueous solution at pH 4 is equal to the value in water. For each other pH, the LIOAS signal from a matched solution of the calorimetric reference BCP was compared to the signal of the BCP solution at pH 4. The sound velocity and the density of the solutions were taken into account in order to calculate the (cpr/b)pH value.16
Results
The UV-vis absorption spectra due to the MLCT transition of Ru(bpy)(CN)42- in aqueous solutions at various HCl concentrations show a marked pH dependence (Figure 1). As the acidity increases, a blue shift of the MLCT band and at least two points at 333 and 378 nm which are almost isosbestic are observed.
The spectral changes have been assigned to the first step ground-state protonation of the complex A2- according to the equilibrium reaction A, with a pKa = 1.8.12
A2- + H+ ![]() AH- where
A2- = Ru(bpy)(CN)42- (A)
AH- where
A2- = Ru(bpy)(CN)42- (A)
The blue shift observed is consistent with the dRu®p*bpy nature of the MLCT transition. This sensitivity to the environment is due to the tendency of the cyanide ligands to interact with the proton through second-sphere type interactions, which results in a lower electron density in the cyanide ligands. In spite of the changes in the absorption spectra with pH, the emission spectra obtained upon excitation in the isosbestic point at 378 nm of Ru(bpy)(CN)42- are practically independent of the pH (Figure 1).
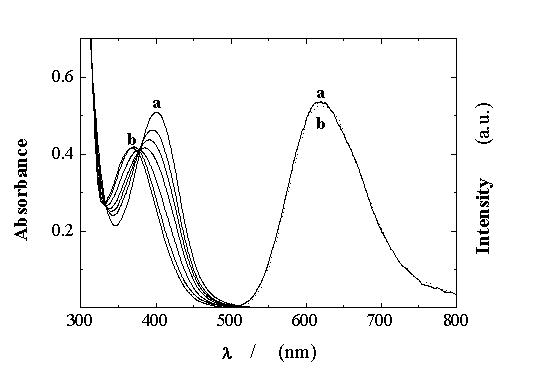
Figure 1. Effect of pH on the UV-vis absorption and emission spectra (lexc = 379 nm) of Ru(bpy)(CN)42- ; pH values between (a) 6.63 and (b) 0.35.
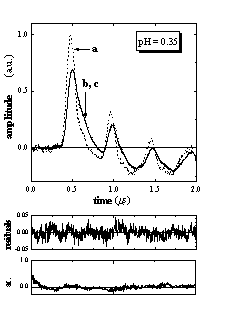
Figure 2 shows the LIOAS signals obtained for Ru(bpy)(CN)42- in aqueous solutions at pH = 0.35 and 6.63, together with those for the calorimetric reference BCP. The shape of the sample signal depends on the pH value. Similar to our previous reports,13,15 satisfactory fits of the LIOAS signals for the sample were obtained using a biexponential decay function (Figure 2). In all cases, the fitting procedure yields two well separated decay times: t1 < 5 ns and t2 between 100 and 110 ns, when all four parameters (two lifetimes and two preexponential factors) are allowed to vary freely. The value of t1 only means that this decay is faster than the time resolution of the experiment. Fixing this parameter at any value between 1 and 10 ns always resulted in a similar value of the associated amplitude of the process. This amplitude ř1 is thus a reliable measure of the prompt processes, i.e., the formation of the triplet MLCT state taking place with an efficiency close to unity (FR»1).13,15,17 The second amplitude ř2 and its respective decay time are assigned to the MLCT decay, i.e., t2 = tMLCT.13,15 In all cases we found ř1 + ř2 = 1 ± 0.05, indicating that all excited MLCT species return to the ground state.
 |
|
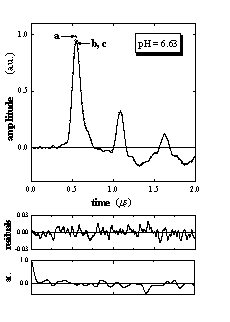
Figure 2. LIOAS signals for (a) the calorimetric reference bromocresol purple (BCP) and for (b) Ru(bpy)(CN)42- upon excitation at 400 nm, at 25 °C and pH = 0.35 and 6.63, together with (c) the fit, residuals, and the autocorrelation waveforms, after deconvolution with a biexponential function describing the pressure time evolution. The curves of sample and fit are indistinguishable. Fit results were at pH = 0.35: ř1 = 0.41, ř2 = 0.63, t2 = 105 ns, c2 1.35 10-4 and at pH = 6.63: ř1 = 1.06, ř2 = -0.08, t2 = 108 ns, c2 = 5.64 10-5.
For the whole pH range studied, the plots of Elř1 and Elř2 vs. cpr/b were linear as predicted by eq. 1 (Figure 3). The intercepts, q1 and q2, are the heat released in the formation and decay of the MLCT state, respectively. The slopes yield the structural volume change associated with these processes, since FR = 1 (vide supra). The experimental data are collected in Table 1.
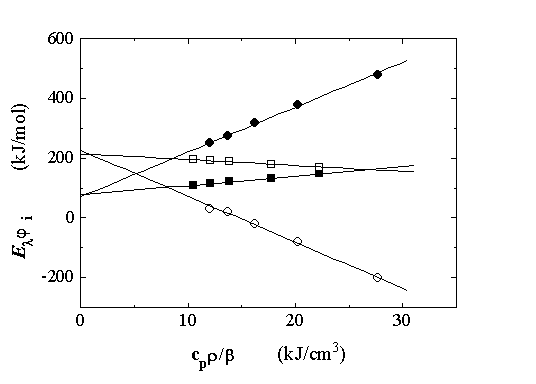
Figure 3. Elř1 and Elř2 vs. cpr/b, associated with the formation (filled symbols) and decay (open symbols) of the MLCT of Ru(bpy)(CN)42- in aqueous solutions at pH = 0.35 (squares) and pH = 6.63 (circles). Temperatures for LIOAS measurements (from left to right): 35, 30, 25, 20, and 15 oC.
Table 1. Prompt (q1) and slow (q2) heat, enthalpy content (DHMLCT), structural volume changes (DVMLCT), and decay times (tMLCT) associated with the metal-to-ligand charge-transfer (MLCT) state of the Ru(bpy)(CN)42- complex in water as a function of the pH.
pH |
q1 (kJ/mol) |
q2 = DHMLCT (kJ/mol) |
El-q1 = DHMLCT (kJ/mol) |
DVMLCT (formation) (cm3/mol) |
DVMLCT (decay) (cm3/mol) |
tMLCT (ns) |
0.35 0.51 0.68 1.02 1.29 1.52 1.70 2.28 2.82 4.30 6.05 6.63 |
77 ± 6 75 ± 7 79 ± 8 73 ± 5 81 ± 10 68 ± 5 61 ± 15 72 ± 10 69 ± 8 66 ± 10 64 ± 9 68 ± 10 |
214 ± 8 214 ± 10 211 ± 10 209 ± 6 204 ± 8 224 ± 15 231 ± 13 226 ± 10 228 ± 10 230 ± 10 230 ± 10 230 ± 10 |
222 ± 6 224 ± 7 220 ± 8 226 ± 5 218 ± 10 231 ± 5 238 ± 15 227 ± 10 230 ± 8 233 ± 10 235 ± 9 231 ± 10 |
+ 3.0 ± 0.3 + 2.9 ± 0.4 + 3.3 ± 0.5 + 3.8 ± 0.3 + 5.3 ± 0.4 + 7.9 ± 0.3 + 9.9 ± 0.9 + 12.8 ± 0.5 + 13.7 ± 0.5 + 15.1 ± 0.5 + 14.7 ± 0.5 + 15.0 ± 0.5 |
- 2.5 ± 0.6 - 2.8 ± 0.5 - 3.8 ± 0.6 - 4.0 ± 0.4 - 5.4 ± 0.4 - 8.8 ± 0.8 - 9.8 ± 0.7 - 13.0 ± 0.5 - 14.0 ± 0.7 - 15.3 ± 0.5 - 15.0 ± 0.5 - 15.1 ± 0.5 |
103 ± 10 105 ± 10 106 ± 15 110 ± 15 108 ± 10 112 ± 10 107 ± 10 106 ± 15 110 ± 10 104 ± 10 110 ± 10 107 ± 10 |
Discussion
The effect of pH on absorption and emission spectra, and on the lifetimes of the excited Ru(bpy)(CN)42- has been already explained by Indelli et al.12 The excited state of protonated Ru(bpy)(CN)42- is remarkably more acid than its ground state, due to the electron transfer from the central metal to the bipyridine ligand which leads to a decrease in electron density at the protonation site. Thus, a fast deprotonation process (reaction B) of the excited state precedes emission.
AH- + hn ® *AH- ® *A2- + H+ (B)
Consequently, throughout the pH range studied in the present work the properties of the excited state, such as emission maxima and tMLCT, are those of the deprotonated complex Ru(bpy)(CN)42-.
The enthalpy content of the MLCT state of Ru(bpy)(CN)42-, DHMLCT, is estimated from the values of q1 and q2 at pH > 3, where ground state protonation does not occur. For the prompt process, DHMLCT is calculated as the energy difference between the excitation energy El and the heat released q1. Since Ru(bpy)(CN)42- has a low fluorescence quantum yield,18 the amount of energy lost by radiation (EemFem < 2 kJ/mol) is negligible compared to the excitation energy El. For this reason, and since the MLCT state of Ru(bpy)(CN)42- fully returns to the ground state in about 110 ns, the heat released in the decay process, q2, equals DHMLCT. The calculated values are listed in Table 1. The values of DHMLCT corresponding to the columns q2 and El-q1 are almost identical at pH > 1.7 and in very good agreement with the value of 228 ± 2 kJ/mol reported for water.13
At lower pH’s an increment of about 26 kJ/mol in DHMLCT is expected due to the blue spectral shift produced by protonation of the ground state. However, at pH < 1.7, q2 < El - q1 and lower than 228 kJ/mol. This should be due to the fact that the hydration of the ionic species produced in reaction B is exothermic.
The values of DVMLCT strongly depend on pH. The observed DVMLCT values for the MLCT formation decrease from +15.0 cm3/mol at pH = 6.63 to +3.0 cm3/mol at pH = 0.35. For the decay process the same change (of values with the opposite sign) is observed.
The values of DVMLCT for the MLCT formation and decay of Ru(bpy)(CN)42- in water solutions15 and in the water pool of reverse micelles13 have been rationalized on the basis of hydrogen bond interactions between the complex and the first solvation shell. Upon MLCT formation the change in the oxidation state from Ru(II) to Ru(III) is accompanied by a strong reduction in the p back-bonding and by a shift in electron density from the cyanide ligands to the central metal. Thus, the hydrogen bonds to the water molecules are expected to be weakened, resulting in an expansion of the solvation sphere. In turn, when the MLCT state decays, the opposite effect occurs and a contraction of the same magnitude is observed. In aqueous solutions, the magnitude of DVMLCT is also dependent on the number of cyano ligands able to interact with the water molecules of the first solvation shell, as has been reported for the homologous derivatives Ru(bpy)(CN)3(CNCH3)- and Ru(bpy)(CN)2(CNCH3)2 , with DVMLCT = 10 and 0 cm3/mol, respectively.15
The value of DVMLCT for the formation of the MLCT of Ru(bpy)(CN)3(CNH)- at pH = 0.35 is smaller than the corresponding value for the homologous anion Ru(bpy)(CN)3(CNCH3)-. A qualitative explanation would be the increment in electrostriction due to the increase in the number of charged particles present in the solution after the prompt deprotonation of the excited state. The electrostriction effect is associated with a contraction of the medium due to the reorganization of the solvent by the electrical field of the ions produced by the process represented in eq. B.
In order to estimate quantitatively this contribution we assume that at pH = 0.35 the Ru(bpy)(CN)42- complex is almost totally monoprotonated. Unfortunately, due to the sequential protonation steps undergone by this complex, at lower pH’s a second protonation starts.12 This is also evidenced by the not totally well defined isosbetic points. However, at 400 nm the diprotonated species does not absorb due to the strong blue shift of the MLCT band,12 and the small fraction present (less than 3 %) will not interfere with the absorption of the monoprotonated species. The observed value of DVMLCT can be taken therefore as the value associated with the monoprotonated form of Ru(bpy)(CN)42-. Similarly, the values of DVMLCT at high pH values, i.e., pH = 6.63, correspond to the fully deprotonated complex. Within this framework and since the excitation wavelength for the LIOAS experiments does not correspond to the isosbestic point for the ground state acid-base equilibrium, the pH dependence of DVMLCT values is given by eq. 2,
DVMLCT pH (cm3/mol) = fAH- DVMLCT pH=0.35 + fA2- DVMLCT pH=6.63 (2)
where fAH- and fA2- are the molar fractions of the monoprotonated and deprotonated forms of Ru(bpy)(CN)42-, respectively. Under our conditions fAH- + fA2- = 1 (vide supra).
Using eq. 2 and the DVMLCT values for the MLCT formation (Table 1), fA2- was calculated and plotted vs. pH (Figure 4), together with the values calculated from the absorbance changes of the deprotonated form of the complex at 475 nm.12 Both titration curves are coincident and a value of pKa = 1.79 ± 0.02 is obtained by using a sigmoidal fitting of the experimental points, in good agreement with reported data.12 This result supports our preliminary assumption that DVMLCT at pH = 0.35 is associated with the monoprotonated species.
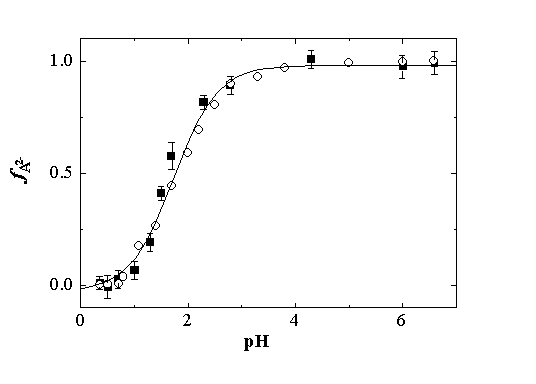
Figure 4. Titrations curves in water, obtained from the fraction of Ru(bpy)(CN)42-, fA2- vs. pH, calculated from (n) volume changes and eq. 2, and (ˇ) absorption changes at 475 nm. The solid line represents a sigmoidal fitting of both sets of experimental data. pKa = 1.79 ± 0.02.
Taking into account reaction B, DVMLCT at pH = 0.35 is expressed by eq. 3 in terms of the individual partial molar volumes
DVMLCT pH= 0.35 = (V*AH--VAH- ) + [(V*A2 - + VH+ ) -V *AH- ]
= VH+ + (V*A2 - -V AH- ) (3)
V*A2- is replaced by the value of DVMLCT at pH values at which ground state protonation does not occur, i.e., DVMLCT pH=6.63 = (V*A2- - VA2- ). Finally, eq. 4 for the partial molar volume of the proton is obtained by rearranging eq. 3,
VH+ = (DVMLCT pH=0.35 - DVMLCT pH=6.63 ) - (VA2- -VAH- ) (4)
In order to use eq. 4, the value of (VA2- -VAH-) must be estimated. In principle, the partial molar volume associated with an ion in solution, Vion, should be the sum of a least two contributions: (i) an intrinsic partial volume, Vint , and (ii) the partial volume due to electrostriction, Velect .19
In our case the changes in the intrinsic volume contribution due to the deprotonation of AH- to A2- can be neglected since both ions should have a similar size. However, the electrostriction contribution induced by the change in the charges of the complex cannot be neglected and may be calculated by using the Drude-Nernst eq. 5,1
 (5)
(5)
where z and r are the charge and the radius of the ion, respectively. The model is relatively simple and assumes that the solvent is a continuum of dielectric constant e, interacting with hard sphere ions only through coulombic forces. The volume change due to electrostriction, Velect, is thus related to the partial derivative ¶ln e/ ¶p. In water at 25 şC the theoretical value of the constant B is 4.175 for r in A. However, experimental results have shown that much higher B values (obtained by semiempirical calculations) are needed to explain the partial molar volume of ions in water.1 Therefore, taking into account only electrostriction effects induced by the change in the charge of the complex, a value (VA2--VAH-) = (-6.9 ± 0.6) cm3/mol is calculated, by using B = 9.620 and r = 4.2 Ĺ in the Drude-Nernst eq. 5. The radius of Ru(bpy)(CN)42- was calculated as ˝(dxdydz)1/3, where d are the diameters along the molecular axes dx = dy = 9.5 Ĺ (NC-Ru-bpy) and dz = 6.4 Ĺ (NC-Ru-CN) as estimated from crystallographic data for [Ru(bpy)3][PF6],21 and for Ru(CN)6Na4.22
The apparent partial molar volume of the proton in water is thus estimated to be (-5.1 ± 0.8) cm3/mol, by using eq. 4 and the DVMLCT values at pH = 0.35 and pH = 6.63.
However, this value is obtained from LIOAS measurements performed in relatively concentrated solutions. The partial molar volume at infinite dilution can then be calculated by applying the extended Debye-Hückel eq. 6 derived by Pitzer,23 valid in the range of concentrations (ionic strengths) used in this work,
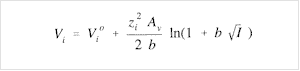 (6)
(6)
where Vi is the apparent partial molar volume of the ionic species expressed as a function of the actual ionic strength, I , of the solution (on molality basis). Vio is the partial molar volume at infinite dilution, zi is the ion charge, b = 1.2 (kg/mol)1/2, and Av = 1.874 cm3 kg1/2 mol-3/2 at 25 °C is the Debye-Hückel limiting slope for the partial molar volume. Thus, the correction by ionic strength effects results in ca. -0.4 cm3/mol for the more concentrated solution (pH = 0.35), lower than the error of the calculated proton partial molar (0.8 cm3/mol).
By convention, V°H+ = 0.0 cm3/mol. Many workers have measured the "absolute" proton partial molar volume using several approaches.10 In all cases negative partial molar volumes have been calculated irrespective of the method, with values ranging between -1 and -8 cm3/mol. For example, in Table XX of ref. 1 a list of V°H+ values in water at 25 °C is given. In a more recent review, a contraction of 5.5 cm3/mol is reported.24 Thus, our result of V°H+= (-5.5 ± 0.8) cm3/mol, corrected for ionic strength, falls within the literature data.1,10 We note that the error of our measurement is smaller than the scatter of the reported data. We also note that although our value depends on B [eq. 5 for the calculation of the term (VA2- -VAH-)] we have used the value recommended for water after a critical analysis of all the data reported in the literature.25
Finally, our values agree also with previous LIOAS data. In the case of pyranine (8-hydroxypyrene-1,3,6-trisulphonate) in water a structural volume change of -6.9 cm3/mol was observed.8 The authors mentioned that this change may be influenced by pyranine anion solvation without elaborating its magnitude. In fact, it is possible to estimate this effect by subtracting the value of V°H+= (-5.5 ± 0.8) cm3/mol from the structural volume change of -6.9 cm3/mol. The difference of -1.4 cm3/mol seems to be a reasonable amount for solvent reorganization due to the charge increase after proton ejection from a bulky anion like pyranine. More recently, Bonetti et. al. 9 calculated a structural volume change of (-5.2 ± 0.2) cm3/mol at low pH for the solvation of the ions (nitronate anion plus the proton) formed after the photoinduced proton ejection of o-nitrobenzaldehyde. The fact that this value coincides with the value of V°H+ calculated in the present report, may be taken as an indication that the volume change contribution due to the formation of the nitronate anion from o-nitrobenzaldehyde is very small, in spite of the internal rearrangement of the molecule taking place after photoexcitation.26
Conclusions
Partial molar volume changes of ionic species photoproduced in dilute aqueous solutions cannot always be measured by conventional methods, and often the linear extrapolation from measurements performed at higher concentrations to infinite dilution is not warranted. LIOAS offers an efficacious alternative in such cases since partial molar volumes are readily calculated from the structural volume changes determined by LIOAS.
It should be possible to extend the principle of the present study of the proton partial molar volume in water to the determination of the partial molar volume of other ions by using LIOAS in combination with a suitable complexation agent photoejecting a particular ion. In particular, proton translocation studies with biological systems (otherwise not photoinducible) could be studied by this technique. In any case, in order to obtain information about the partial molar volume contribution of the ejected ions into the solution, the intrinsic and electrostrictive volume effects of the produced counterion must be carefully evaluated.
Acknowledgements. The able technical assistance of Sigrid Pörting-Russell, Carmen Berling and Dagmar Lenk is greatly appreciated. We thank Professor Franco Scandola who provided us the samples of K2 Ru(bpy)(CN)42- and Professor Kurt Schaffner for his constant interest in and support of this work.
References and Notes
[1] F.J. Millero, Chem. Rev. 71 (1971) 147-176.
[2] W.J. le Noble, H. Kelm, Angew. Chem. 78 (1980) 841-856.
[3] R. Van Eldick, T. Asano, J.L. le Noble, Chem. Rev. 89 (1989) 549-688.
[4] S.E. Braslavsky, G.E. Heibel, Chem. Rev. 92 (1992) 1381-1410.
[5] J.I. Kim, R. Stumpe, R. Klenze, Topics Curr. Chem. 157 (1990) 129-179.
[6] J.B. Callis, W.W. Parson, M. Gouterman, Biochim. Biophys. Acta 267 (1972) 348-362.
[7] C.D. Borsarelli, H. Corti, D. Goldfarb, S.E. Braslavsky, J. Phys. Chem. A 101 (1997)
7718-7724.
[8] J.R. Small, E. Kurian, Spectroscopy 10 (1995) 27-33.
[9] G. Bonetti, A. Vecli, C. Viappiani, Chem. Phys. Lett. 269 (1997) 268-273.
[10] G. Hefter, Y. Marcus, J. Solut. Chem. 26 (1997) 249-266.
[11] S. Bickel-Sandkötter, M. Dane, W. Gärtner, Arch. Microbiol. 166 (1996) 1-11.
[12] M.T. Indelli, C.A. Bignozzi, A.M. Marconi, F. Scandola in Photochemistry and
Photophysics of Coordination Compounds, H. Yersin, A. Vogler, eds., p. 159, Springer-
Verlag, Berlin (1987).
[13] C.D. Borsarelli, S.E. Braslavsky, J. Phys. Chem. B 101 (1997) 6036-6042.
[14] J.E. Rudzki, J.L. Goodman, K.S. Peters, J. Am. Chem. Soc. 107 (1985) 7849-7854.
[15] J.L. Habib Jiwan, B. Wegewijs, M.T. Indelli, F. Scandola, S.E. Braslavsky, Recl. Trav.
Chim. Pays-Bas 114 (1995) 542-548.
[16] M.S. Churio, K. Angermund, S.E. Braslavsky, J. Phys. Chem. 98 (1994) 1776-1782.
[17] F. Scandola, M. T. Indelli, Pure Appl. Chem. 60 (1988) 973-980.
[18] C.A. Bignozzi, C. Chiorboli, M.T. Indelli, M.A. Rampi Scandola, G. Varani,
F. Scandola, J. Am. Chem. Soc. 108 (1986) 7872-7873.
[19] P. Pollmann, D. Rehm, A. Weller, Ber. Bunsenges. Phys. Chem. 79 (1975) 692-696.
[20] Average value for the value in water B = 9.6 ± 1.5, from reference 1. Page 171
[21] D.P. Rillema, D. S. Jones, H.A. Levy, J. Chem. Soc. Chem. Comm. (1979) 849-851.
[22] L.A. Gentil, A. Navaza, J.A. Olabe, Inorg. Chim. Acta 179 (1991) 89-96.
[23] K.S. Pitzer, J. Phys. Chem. 77 (1973) 268-277.
[24] Y. Markus, Biophys. Chem. 51 (1994) 111-127.
[25] F. Millero in Water and Aqueous Solutions, R.A. Holmes, ed., Wiley-Interscience, New
York (1972).
[26] R.A. McClelland, S. Steenken, Can. J. Chem. 65 (1987) 353-356.