THE EFFICIENCY OF SINGLET OXYGEN GENERATION BY ALLOXAZINE AND LUMICHROME
M. Sikorski a, E. Sikorska b, D.R. Worrall c and F. Wilkinson c
a Faculty of Chemistry, A. Mickiewicz University, 60-780 Poznan, Poland b Faculty of Commodity Science, Poznan University of Economics, 60-967 Poznan, Poland c Department of Chemistry, Loughborough University, Loughborough, Leicestershire LE11 3TU, England
Abstract
The efficiencies of the photosensitised production of singlet oxygen by alloxazine and lumichrome have been determined in acetonitrile. For these compounds the measurements have demonstrated that the efficiency of singlet oxygen production from the triplet state is high. In addition, the lifetimes of the singlet and triplet states and some spectral properties of the compounds in acetonitrile are also reported.
Introduction
Alloxazines are products of the
decomposition of biologically important flavins and can be associated with flavins in many
organisms. The structure and abbreviations of alloxazine and lumichrome are presented in
Figure 1. 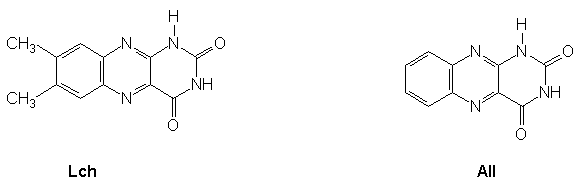 Figure
1.- Structures of lumichrome, Lch, and alloxazine, All.
Figure
1.- Structures of lumichrome, Lch, and alloxazine, All.
Differently substituted alloxazines, mainly lumichromes, have been found in biological material [1]. For example, a lumichrome product of the photodecomposition of riboflavin (vitamin B2) has been found in milk and in dairy products [1,2]. In this context information regarding the photochemistry of alloxazines is of importance. Of particular interest is information about reactions with oxygen and about the production of singlet oxygen. There is strong evidence for the involvement of singlet oxygen in many damaging photooxidations in biological systems. For example singlet oxygen reacts with a wide variety of compounds containing carbon-carbon double bonds and conjugated double bonds, which are structural attributes of all biologically important substrates [3,4,5]. A physical property of singlet oxygen which should be noted is that its radiative relaxation to its ground state can be monitored at 1270 nm, this and non-radiative relaxation channels competing with reactions of singlet oxygen with oxidizable substrates.
In this paper we present some photophysical properties of alloxazine and lumichrome with special emphasis on the study of the efficiency of singlet oxygen generation in acetonitrile solutions. To learn more about the interaction between alloxazines and oxygen in acetonitrile solution we have determined the kinetic properties of the studied compounds in excited singlet and triplet states in the presence and in the absence of oxygen. Additionally we have measured the phosphorescence from singlet oxygen generated from oxygen quenching of the excited states of these compounds monitored at 1270 nm.
Experimental
The measurements of transient absorption were performed using a nanosecond laser flash photolysis system with right angle geometry which has been described in detail elsewhere [6,7]. In brief, the excitation source was the third (355 nm) harmonic of a JK2000 Q-switched Nd:Yag laser (Lumonics) with an energy of 20 mJ and a pulse width (FWHM) of 30 ns. The analysing source was a 300 W xenon arc lamp (LOT Oriel Ltd) used in combination with a monochromator (Applied Photophysics Ltd, F/3.4 grating) for collecting the analysing light which was detected by a R28 photomultiplier (Hamamatsu).
Emission from singlet oxygen following laser excitation was detected by time resolved spectroscopy as described previously [8]. Singlet oxygen was detected by monitoring the 0,0 vibronic band of the phosphorescence centered at 1270 nm using a Judson germanium photodiode coupled to a Judson PA100 preamplifier. The phosphorescence was detected at right angles to the exciting beam. The intensity of singlet oxygen phosphorescence (I0) at time t=0 was obtained by fitting the decay curve to a single exponential function. For each sample I0 values were plotted against relative laser intensity to obtain plots which were linear below 0.5 mJ per pulse. Air-equilibrated solutions of the alloxazine and lumichrome were optically matched (+/- 0.003 absorbance units) at the excitation wavelength (355 nm) to a standard reference solution. Solutions in acetonitrile were prepared in 1-cm-square cells with absorbance at 355 nm 0.6 for lumichrome and 0.3 for alloxazine. The quanutm yield of photosensitised production of singlet oxygen, fD, values were determined relative to the slope obtained for the plot of I0 vs. relative laser intensity for acridine in air saturated acetonitrile for which a value of 0.82 has been measured [9].
The fluorescence decay curves were measured using a time-correlated single-photon counting method on the commercially available IBH model 5000U fluorescence lifetime spectrometer. Time-resolved fluorescence data were fitted to single exponential by an iterative convolution method employing a least-squares fitting procedure.
Fluorescence spectra and absorption spectra were recorded using a Fluoromax spectrofluorometer (SPEX industries) and a Philips PU8800 spectrophotometer respectively. The acetonitrile as a solvent was spectrophotometric grade from Aldrich and was used as received. Samples were prepared in quartz emission cells (1cm2).
Results and discussion
Interest in the photochemistry of alloxazines began in 1966 when the proton transfer reaction in alloxazines was discovered [10-13]. However, much of the photophysical data of alloxazines are still unknown. In this paper we present some spectral and photophysical data for the singlet and triplet states of the two examined compounds, alloxazine and lumichrome, in acetonitrile solution.
Table 1 gives a summary of the photophysical
parameters of the singlet states of the studied compounds.
| Compound | lmax(2nd) | lmax(1st) | lfl | ffl | tfl |
| (nm) | (nm) | (nm) | (ns) | ||
| alloxazine | 320 | 372 (6600) | 432 | 0.0093 | - |
| lumichrome | 334 | 380 (8300) | 439 | 0.0280 | 0.7 |
Table 1: Spectral data for alloxazine and lumichrome in acetonitrile; the position of two long-wavelength bands in the absorption spectra of alloxazines - lmax(1st), lmax(2nd), in brackets are given extinction coefficients. The position of the fluorescence emission spectra - lfl, the quantum yields, ffl, and lifetime of fluorescence - tfl, are also given.
For both alloxazine and lumichrome, there are two maxima in the absorption spectra between 280 and 500 nm. As predicted from theoretical calculations, these two long-wavelength bands in the absorption spectra of alloxazines reflect two independent p -p * transitions [11,14,15]. The numbering of bands - 1st, 2nd, refers to the number of obviously different absorption bands and not necessarily to the true sequence of states. The positions of these maxima are listed in Table 1, where also the extinction coefficients are given. The absorption spectrum for lumichrome is presented in Figure 2.
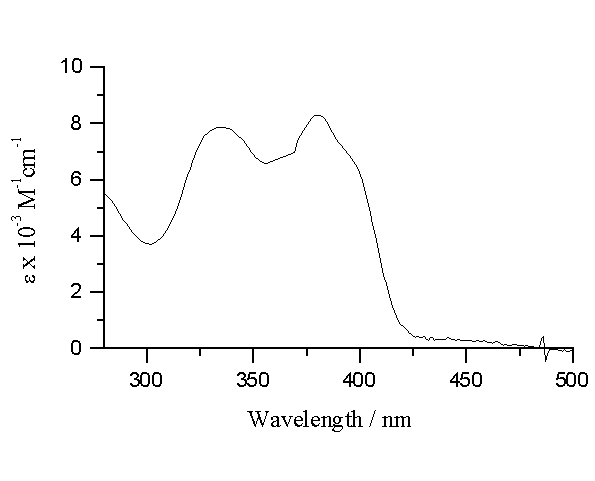
Figure 2.- Ground state absorption spectrum
of lumichrome in acetonitrile.
The fluorescence emission spectra of alloxazine and lumichrome have a single band with a maximum at 432 nm and 439 nm respectively. The positions of maxima are listed in Table 1, where also the quantum yields and lifetimes of fluorescence are given. A representative fluorescence spectrum of alloxazine in acetonitrile is presented in Figure 3.
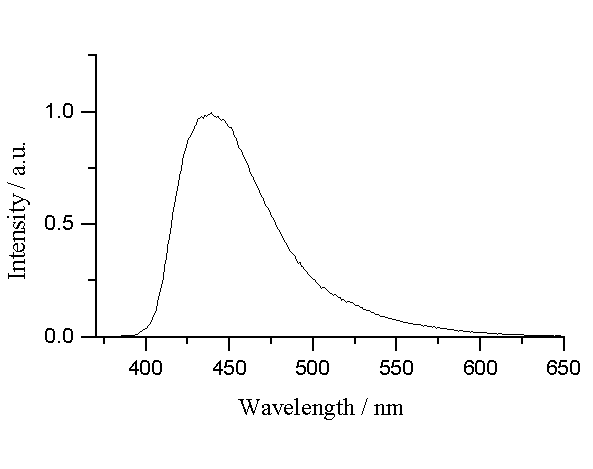
Figure 3.- Fluorescence spectra of lumichrome in acetonitrile at room temperature. The excitation wavelength used for these spectra was 355 nm.
The photophysical properties for alloxazine and lumichrome reported by us in acetonitrile are similar to those reported previously by some from us for 6-methylalloxazine, 6MAll, 7-methylalloxazine, 7MAll, 8-methylalloxazine, 8MAll, 9-methylalloxazine, 9MAll, when 1,2-dichloroethane were used as a solvent [16], including the position of two long-wavelength bands in the absorption spectra of alloxazines - lmax(1st), lmax(2nd), the position of the fluorescence emission spectra of alloxazines - lfl, the quantum yields, ffl, and lifetime of fluorescence - tfl. The lifetimes reported previously in 1,2-dichloroethane are, [16]; 0.87, 0.49, 0.32 and 0.74 ns for 6-methylalloxazine, 6MAll, 7-methylalloxazine, 7MAll, 8-methylalloxazine, 8MAll, 9-methylalloxazine, 9MAll respectively and are similar to those determined by us for lumichrome in acetonitrile. Generally the lifetime of singlet states of alloxazines is very short and varies depending on the position of methyl substituent [16].
The lifetime of singlet state of alloxazine is short and the fact that the fluorescence quantum yields are insensitive to oxygen concentration in acetonitrile when comparing degassed and air equilibrated samples confirms that no oxygen quenching of this state takes place. However we have performed experiments at higher oxygen concentrations to obtain the rate constant of quenching of the singlet state of alloxazines by oxygen in acetonitrile. These rate constants kS[O2] were obtained from the Stern-Volmer equation by measuring the quantum yields of fluorescence as a function of oxygen concentration. The value of kS[O2] was calculated using the lifetime of singlet state of lumichrome presented in Table 1. From these experiments we calculated for lumichrome the value kS[O2] = 6.4 ´ 109 M-1 s-1. Finally we can assume that excited singlet state of lumichrome is not effectively quenched by oxygen in air equilibrated acetonitrile and as consequence can not give the rise to singlet oxygen after the direct excitation to the singlet state at 355 nm.
Transient absorption spectra of the excited triplet state of alloxazines have drawn less attention than have the photophysical properties of alloxazines in their singlet states. To our knowledge no data about triplet-triplet absorption spectra of alloxazines have been reported in acetonitrile. Upon laser excitation at 355 nm, alloxazines in acetonitrile produce transient species that decay on a microsecond time scale. The representative transient absorption spectra for lumichrome, Lch, are
presented in Figure 4.
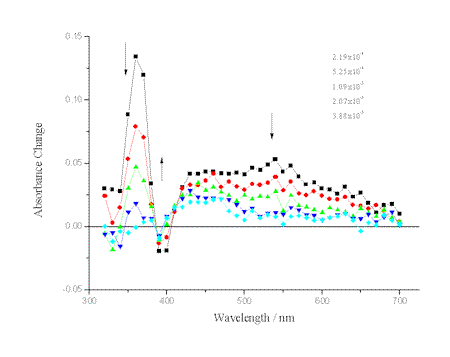
Figure 4.- Transient absorption spectra of lumichrome in acetonitrile at room temperature. The numbers refer to the time after laser excitation at 355 nm. The OD at 355 nm was 0.3.
As shown for Lch, a negative absorbance change at
around 390 nm is observed because of ground-state depletion. The spectra in Figure 4 are
similar to those previously reported for lumichrome and alloxazine in different solvents
[17-19]. The triplet photophysical data for the two studied compounds in acetonitrile are
summarised in Table 2. For lumichrome and alloxazine at least two species were previously
postulated in the transient absorption spectra in aqueous solutions at various values of
pH and also in ethanol solutions [17,19-21]. A detailed discussion of the kinetics of
transient decay will be presented in a separate paper. For the discussion about the
interaction of alloxazines with oxygen we can assume on the basis of previous studies that
the maximum at about 490 nm for alloxazines and about 540 nm for lumichromes can be
assigned as the triplet state, see references [17,19]. The lifetimes of triplet state
presented in Table 2 have been calculated from their decay using the wavelength of 3rd
maximum listed also in Table 2. For example the previously reported lifetime of the
triplet state in aqueous solution at pH 2.2 are 12.0 ms for Lch, [17] and 9.4 ms for alloxazine, [17]; in addition for alloxazine in
ethanol, the value of 13 ms was also reported, [19]. The lifetimes reported by
others correspond very well with these presented by us in Table 2.
| Compound | tT/ms | fT | |||
| (nm) | (nm) | (nm) | |||
| alloxazine | 360 | 430 | 500 | 10 | 0.45** |
| lumichrome | 360 | 450 | 540 | 11 | 0.71** |
Table 2. The triplet photophysical data
for alloxazine and lumichrome in acetonitrile. The position of maxima in transient
absorption spectra (![]() ,
, ![]() ,
, ![]() ), the lifetime of triplet state, tT/ms, and the quantum yields of intersystem crossing, fT are presented.
), the lifetime of triplet state, tT/ms, and the quantum yields of intersystem crossing, fT are presented.
** in neutral aqueous solution - from Ref. [17]
In this work we measured the emission of singlet
oxygen at 1270 nm, which is highly specific to the O2 (1D g) ® O2 (3ĺg-)
transition, following laser excitation at 355 nm of air-equilibrated acetonitrile
solutions of alloxazine and lumichrome. The emission intensity at 1270 nm increased in a
samples with higher concentration of oxygen. The emission was diminished by bubbling N2
into the solution for a few minutes. Also the lifetimes of the emission recorded at 1270
nm and presented in Table 3 are characteristic for the lifetimes of singlet oxygen in
acetonitrile solutions[3]. These observations confirm that all the compounds used in this
study acted as photosensitizers of singlet oxygen, and that 1O2 was
responsible for the emission at 1270 nm.
| Compound | fD | tD /m s |
| alloxazine | 0.36 | 70 |
| lumichrome | 0.73 | 72 |
Table 3. Quantum yields of photosensitised production of singlet oxygen, fD , and singlet oxygen lifetimes, tD , in air-equilibrated acetonitrile solutions.
The values of the quantum yield of formation of singlet oxygen by sensitisation, fD, were determined relative to the slope obtained for the plot of I0 vs relative laser intensity for acridine in air saturated acetonitrile for which a value of 0.82 has been measured [9], as described in experimental section. A summary of the values of fD deteremined, as well as values of singlet oxygen lifetimes, tD , in air-equilibrated acetonitrile solutions are given in Table 3.
Generally the quantum yield of formation of singlet oxygen by sensitisation, fD , is given by the sum of the contributions due to oxygen quenching of the lowest excited singlet state ( S1 ) and the lowest excited triplet state ( T1 ) of the oxygen sensitiser:
fD = fD( S1 ) + fD( T1 ) (1)
If fDs and fDT represent the fraction of S1 and T1 states quenched by oxygen which give rise to singlet oxygen ( 1D g ) it follows that:
fD = fDsPs + fTfDTPT (2)
where PT and PS represent the fraction of triplet and singlet states quenched by O2 respectively and fT is the quantum yield of the triplet state formation. The lifetimes of the singlet states of alloxazine and lumichrome obtained in acetonitrile are of order of hundreds of picoseconds (see Table 1), and as discussed previously there is no oxygen quenching of the excited singlet states in air equilibrated solution. In addition the energy gap between S1 and T1 state is to small for the process S1 + 3O2 ® T1 + 1O2 to occur.
Moreover it is well known that for some alloxazines fT is high, Table 2. Finally we can assume that the quantum yield of singlet oxygen observed in this work is the sole result of triplet state quenching,
fD = fTfDTPT (3)
In Table 3 are presented quantum yields of photosensitised production of singlet oxygen, fD , in air-equilibrated acetonitrile solutions. These results demonstrate how good the alloxazine and lumichrome are at photosensitising of singlet oxygen. The presented values of fD are high, with the exception of the quantum yield of singlet oxygen for alloxazine. The latter is probably due to the effect of a smaller value of the quantum yield of intersystem crossing. The lifetimes of singlet oxygen reported by us in Table 3 are in good agreement with expected values in acetonitrile [3]. These results indicate that alloxazines, as relatively photostable products of photolysis of flavins, may act as efficient sensitisers of singlet oxygen. Therefore they may be responsible for some oxidation reactions occurring in biological materials, since for example as was mentioned previously the presence of lumichrome as a product of the photodecomposition of vitamin B2 has been found in milk [2].
Acknowledgement
Financial support from The Royal Society/Central & East European Postdoctoral Fellowship, The Foreign & Commonwealth Office to M. S and The British Council and KBN for the grant to F.W and M.S are gratefully acknowledged.
References
| [1] J. Chastain and D. B. McCormick, in F. Muller (ed.), Chemistry and Biochemistry of Flavoenzymes, vol 1. CRC Press, Boston, 1991 p.196. | |
| [2] T. Toyosaki, A. Hayashi, Milchwissenschaft
- Milk Science International, 48 (1993) 607. [3] F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 24 (1995) 663. [4] F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 22 (1993) 113. [5] E. A. Lissi, M. V. Encias, E. Lemp and M. A. Rubio, Chem. Rev., 93 (1993) 699-723. [6] F. Wilkinson, D. R. Worrall, D. J. McGarvey, A. Goodwin and A. Langley, J. Chem. Soc., Faraday Trans., 89 (1993) 2385. [7] M. Sikorski, I. V. Khmelinskii, W. Augustyniak and F. Wilkinson, J. Chem. Soc., Faraday Trans., 92 (1996) 3487. [8] F. Wilkinson, D. J. McGarvey and A. F. Olea, J. Phys. Chem., 98 (1994) 3762. [9] R. W. Redmond and S. E. Braslavsky, In Photosensitization; G. Moreno, R. H. Pottier, T. G. Truscott, Eds.; NATO ASI Series, Vol H15, Springer; Berlin, 1988; p 93. [10] J. Koziol, Photochem. Photobiol., 5 (1966) 41. [11] M. Sun, T. A. Moore and P. S. Song, J. Am. Chem. Soc., 94 (1972) 1730. [12] P. S. Song, M. Sun, A. Koziolowa, and J. Koziol, J. Am. Chem. Soc., 96 (1974) 4119. [13] J. Koziol, Photochem. Photobiol., 5 (1966) 41. [14] J. Komasa, J. Rychlewski and J. Koziol, J. Mol. Struct. (Theochem.), 170 (1988) 205. [15] H. Szymusiak, J. Konarski and J. Koziol, J. Chem. Soc., Perkin Trans., 2 (1990) 229. [16] E. Sikorska and A. Koziolowa, J. Photochem. Photobiol., A. Chemistry, 95 (1996) 215. [17] M. S. Grodowski, B. Veyret and K. Weiss, Photochem. Photobiol., 26 (1977) 341. [18] A. Koziolowa, Photochem. Photobiol., 29 (1979) 459. [19] R. H. Dekker, B. N. Srinivasan, J. R. Huber and K. Weiss, Photochem. Photobiol., 18 (1973) 457. [20] R. R. Düren, R. H. Dekker, H. C. A. Van Beek and W. Berends, Photochem. Photobiol., 23 (1976) 163. [21] P. F. Heelis and G. O. Phillips, J. Phys. Chem., 89 (1985) 770. |