Mimicry of carotenoid photoprotection in artificial photosynthetic reaction centers: Triplet-triplet energy transfer by a relay mechanism
Devens Gust,* Thomas A. Moore,* Ana L. Moore,* Darius Kuciauskas, Paul A. Liddell, and Brian D. Halbert
Center for the Study of Early Events in Photosynthesis, Department of Chemistry and Biochemistry, Arizona State University, Tempe, AZ 85287-1604
Abstract
Two artificial photosynthetic reaction centers consisting of a porphyrin (P) covalently linked to both a carotenoid polyene (C) and a fullerene derivative (C60) have been prepared and found to transfer triplet excitation energy from the fullerene moiety of C-P-3C60 to the carotenoid polyene, yielding 3C-P-C60. The transfer has been studied both in toluene at ambient temperatures and in 2-methyltetrahydrofuran at lower temperatures. The energy transfer is an activated process, with Ea = 0.17 eV. This is consistent with transfer by a triplet energy transfer relay, whereby energy first migrates from C-P-3C60 to the porphyrin, yielding C-3P-C60 in a slow, thermally activated step. Rapid energy transfer from the porphyrin triplet to the carotenoid gives the final state. Triplet relays of this sort have been observed in photosynthetic reaction centers, and are part of the system that protects the organism from damage by singlet oxygen, whose production is sensitized by chlorophyll triplet states. These triads are unique in that they also demonstrate stepwise photoinduced electron transfer to yield long-lived C· + -P-C60· - charge-separated states. Electron transfer occurs even at 10 K. Charge recombination of C· + -P-C60· - yields 3C-P-C60, rather than the molecular ground state. These photochemical events are reminiscent of photoinduced electron transfer in photosynthetic reaction centers.
________________________________________________________________________
1. Introduction
Artificial photosynthetic reaction
centers typically abstract some aspects of natural photosynthetic function and embody them
in simpler, synthetic systems for detailed study. While many attempts to prepare
artificial reaction centers have focused on photoinduced electron transfer,[1-8] which is at the heart of photosynthesis, other features of
the natural process are also important. For example, components of the photosynthetic
apparatus are subject to photodamage, especially at high light levels, and organisms have
developed various photoprotective strategies to mitigate such destruction.[9-17] In this report, we discuss mimicry of one such strategy.
One process leading to photodamage is singlet oxygen sensitization.
Normal excitation of the special pair of bacteriochlorophylls in bacterial reaction
centers is followed by photoinduced electron transfer to generate a bacteriopheophytin
radical anion. This anion in turn reduces a quinone moiety QA, which passes an
electron on to a second quinone acceptor QB. The oxidized special pair is
reduced by a cytochrome, regenerating the photocatalyst. After a second photoinduced
electron transfer, QB is fully reduced to the hydroquinone form, leaves the
reaction center, and is replaced by a fresh QB molecule. Under conditions of
high light, the special pair can receive excitation energy from the antenna system and
carry out the initial electron transfer process before the quinone portion of the electron
transport chain is regenerated. Under such conditions, the charge-separated state
comprising the special pair radical cation and bacteriopheophytin radical anion cannot
evolve in the normal way, and instead recombines to generate the triplet state of the
special pair.
Triplet states of chlorophylls are long-lived, energetic species that
can react with other molecules to cause photodamage. A major mechanism for such damage is
sensitization of singlet oxygen. This sensitization is an energy transfer process that
occurs by an electron-exchange mechanism, requiring diffusion-induced collisions between
the chlorophyll triplet and ground-state oxygen. Singlet oxygen is an extremely reactive
species that, left unchecked, can devastate the organism, causing its demise.
Photosynthetic organisms have therefore developed photoprotective strategies. A major one
involves the quenching of chlorophyll triplet states by carotenoid polyenes. The triplet
energy of carotenoids (~0.63 eV[18]) is much below that of
most chlorophylls, and also below the energy of singlet oxygen (0.98 eV). Thus, if a
carotenoid can quench the chlorophyll triplet state rapidly enough to compete successfully
with oxygen for this species, no singlet oxygen will be produced. Bacterial reaction
centers contain a carotenoid polyene, and photoprotection by this mechanism has been
observed in them.
Electron exchange energy transfer requires orbital overlap, and rapid
transfer thus requires that the donor and acceptor be in van der Waals contact, or
otherwise coupled relatively strongly. However, X-ray diffraction studies of bacterial
reaction centers have revealed that the carotenoid is relatively far from the special
pair, which is the chlorophyll triplet state of lowest energy in the reaction center. In
fact, it is located in van der Waals contact with the "accessory"
bacteriochlorophyll on the inactive branch of the reaction center.[19-21]
How then does the carotenoid quench the triplet state of the special pair? It has been
proposed that the process involves a triplet relay.[22-24]
Triplet energy is transferred from the special pair to the adjacent bacteriochlorophyll in
a thermally-activated step, and the resulting bacteriochlorophyll triplet is rapidly
quenched by the carotenoid. This arrangement has the advantage of ensuring photoprotection
while preventing undesirable reactions such as electron transfer between the oxidized or
excited special pair and the polyene.
In this paper, we report the observation of a triplet relay mechanism
in a pair of carotene (C) porphyrin (P) fullerene (C60) triad artificial
reaction centers (structures 1 and 2).
2. Results
2.1. Photochemistry in polar solvents
The preparation and the photochemistry of
triad 1 in polar solvents has been previously reported,[25]
and will be summarized here. The absorption spectrum of 1 in
2-methyltetrahydrofuran solution is essentially a linear combination of the spectra of
model carotenoporphyrin 3 and fullerene[26] 4,
with maxima at 409, 450 (sh), 475, 506, 575, 630 and 705 nm. Porphyrin 5 has
fluorescence maxima at 630 and 699 nm, whereas fullerene 4 has maxima at 714 and
~800 nm.
The fluorescence emission of 1 features spectral shapes
characteristic of both the porphyrin and the fullerene components, but is strongly
quenched relative to that of the model compounds. Time resolved fluorescence measurements
were performed on ~1 X 10-5 M solutions of 1 in 2-methyltetrahydrofuran
using the single photon timing technique. Global analysis (l ex
= 600 nm) of decays at 772, 787 and 795 nm, where emission is mainly from the fullerene,
yielded one significant exponential decay component of 0.032 ns for C-P-1C60.
Excitation at 590 nm, where the porphyrin is the major absorber, gave porphyrin
fluorescence decays in the 625 - 640 nm region whose global analysis yielded one
significant lifetime of 0.010 ns for C-1P-C60. This approaches the
time resolution of the spectrometer. The singlet-state lifetimes of 3 and 4
are 7.22 ns and 1.35 ns, respectively, and the short lifetimes for 1 are thus
consistent with the strong steady-state fluorescence quenching.
By analogy with other porphyrin-fullerene dyads,[27-29] the very short lifetime of C-P-1C60
is interpreted in terms of photoinduced electron transfer from the porphyrin to yield C-P·
+ -C60· - according to step 2 in Figure 1, which shows the relevant transient states of 1 and
their interconversion pathways. The short lifetime of C-1P-C60 is
consistent with decay by a combination of photoinduced electron transfer to give C-P·
+ -C60· - (step 3) and singlet-singlet energy
transfer yielding C-P-1C60 (step 1). Indeed, transient absorption
studies of a porphyrin-fullerene dyad closely related to 1 on the subpicosecond
time scale have demonstrated formation of the P· + -C60· -
charge-separated state.[30]
The C-P· + -C60· - charge-separated
state could evolve by electron transfer from the carotenoid (step 4 in Figure 1) to give C· + -P-C60· -
. This possibility was investigated by nanosecond transient absorption spectroscopy.
Excitation of 1 in deoxygenated 2-methyltetrahydrofuran led to the observation of a
strong carotenoid radical cation transient absorption at ~940 nm, indicative of C· +
-P-C60· - . The charge-separated state has a lifetime of
170 ns and is formed with an overall quantum yield of 0.14. Interestingly, the decay of
the carotenoid radical cation is correlated with the growth of an absorption band at 550
nm (t = 170 ns) which is characteristic of the carotenoid
triplet state. The quantum yield of 3C-P-C60 is 0.13, and its
(oxygen sensitive) lifetime is 4.9 m s. Thus, 3C-P-C60
is formed by radical pair recombination of C· + -P-C60· - (step
9), and recombination to the ground state by step 8 is minimal.
Triad 1 in 2-methyltetrahydrofuran thus demonstrates two-step
photoinduced electron transfer to generate a long-lived, energetic charge-separated state,
mimicking the photoinduced electron transfer processes observed in natural reaction
centers. In toluene solution, however, the behavior is quite different.
2.2. Photochemistry in toluene
The absorption spectrum of
triad 1 in toluene is similar to that in 2-methyltetrahydrofuran, with maxima at
412, 455 (sh), 485, 510, 575, 628, and 706 nm. Time-resolved emission studies[25] have shown that C-1P-C60 decays with a
time constant of only 12 ps. Under these conditions, the lifetime of the singlet state of
model carotenoporphyrin 3 is 7.34 ns. The fullerene emission in the 800-nm region
grows in with a time constant of 12 ps, signaling singlet-singlet energy transfer from the
porphyrin to the fullerene moiety. The porphyrin is acting as an antenna for the
fullerene, whose extinction coefficient in the visible is quite small. The fluorescence
emission from the fullerene component of triad 1 in toluene decays with a lifetime
of 1.52 ns. The corresponding lifetime for model fullerene 4 in this solvent is
1.40 ns. Thus, in toluene, C-P-1C60 is not quenched relative to the
model compound.
More information about the fate of the excited states of 1 in
toluene was obtained from transient absorption studies. Samples were excited at 590 nm,
where most of the absorption is due to the porphyrin moiety, with ~200 fs, 30 m J pulses and interrogated with pulses from a continuum-generated
white-light probe beam. No transients characteristic of the carotenoid radical cation in
the 950-nm region or of the fullerene radical anion in the 1050-nm region were observed.
The fluorescence and picosecond absorption results indicate that in
toluene, C-1P-C60 decays entirely via singlet-singlet energy
transfer to the fullerene to yield C-P-1C60 (step 1 in Figure 1), and that no photoinduced electron transfer occurs from
either of these two excited singlet states. The unperturbed lifetime of C-P-1C60
in this solvent, coupled with the fact that the yield of intersystem crossing to the
triplet state in fullerenes is essentially unity, suggests that the only significant
product of decay of the excited porphyrin and fullerene singlet states of 1 in
toluene should be the fullerene triplet state, C-P-3C60. Transient
absorption studies on the ns time scale were undertaken to investigate this possibility.
Excitation of an argon purged, ~1 X 10-4 M solution of 1 in toluene at
300 K with a 5-ns laser pulse at 590 nm gave rise to a transient absorption with a maximum
at ~550 nm (Figure 2). This transient grows in after the laser
flash with a time constant of 85 ns (Figure 3) and decays with a
lifetime of ~4.2 m s, which decreases when oxygen is admitted
to the cuvette. This transient is assigned to the carotenoid triplet state, 3C-P-C60,
by comparison with spectra of related compounds.[31] The yield
of 3C-P-C60 was determined by the comparative method using the meso-tetraphenylporphyrin
triplet state (f T = 0.67, (e
T-e G)440 = 6.8 X 104
L mol-1cm-1) as the standard and (e T-e G) = 1.4 X 105 L mol-1 cm-1
for the carotenoid. The yield was unity, within experimental error.
Thus, excitation of 1 in toluene is expected to produce the
fullerene triplet state, but the species observed by nanosecond transient absorption is
the carotenoid triplet state. What is the genesis of this state? It cannot involve
intersystem crossing in the carotenoid. The yield of this process in carotenoid polyenes
of this type is essentially zero, the carotenoid moiety is not appreciably excited at 590
nm, and the rise time is inconsistent with this mechanism. The triplet also cannot arise
from the recombination of the C· + -P-C60· - charge-separated
state, as was observed in 2-methyltetrahydrofuran, because no evidence for formation of a
charge-separated state in toluene was found for 1 or for a related
porphyrin-fullerene dyad, as discussed above. It seems likely that the carotenoid triplet
state is produced via triplet-triplet energy transfer from the fullerene triplet state,
formed through normal intersystem crossing of C-P-1C60.
The fullerene triplet state of 4 has an absorption maximum at
~690 nm.[28,32] Under the experimental conditions used for
observation of 3C-P-C60 in 1, the extinction coefficients of
the carotenoid and fullerene triplet states at 690 nm coupled with the recovery of the
detection system from fluorescence-induced saturation at 690 nm precluded observation of
the decay of C-P-3C60. Therefore, the assumption that
triplet-triplet energy transfer is the mechanism of formation of 3C-P-C60
could not be verified.
Consequently, the spectroscopic properties of a second, closely-related
fullerene-containing triad, 2, have been investigated.[33]
Excitation of this triad in toluene solution results in singlet-state photophysics
essentially identical to those for 1, as is expected given the similar structures
of the porphyrin and fullerene moieties. Transient absorption studies on the picosecond
time scale similar to those described above again showed no evidence for formation of
carotenoid radical cations or fullerene radical anions. Thus, photoinduced electron
transfer does not occur. Nanosecond transient absorption studies in argon-purged toluene
similar to those described above with excitation at 625 nm led to the observation of the
carotenoid triplet absorption with a maximum at 550 nm. The triplet decays with a lifetime
of 4.6 m s in the deaerated solvent, and the lifetime decreases
when oxygen is admitted to the cuvette. The quantum yield of the carotenoid triplet state
is unity, based on 590-nm excitation, just as is the case for triad 1. The 550-nm
carotenoid triplet absorption of triad 2 has a rise time of 110 ns (Figure 3), which is similar to the 85 ns rise time observed for
triad 1.
Nanosecond transient absorption measurements of 2 at 690 nm
revealed an absorption corresponding to the fullerene triplet state (l
max ~690 nm [28]). The transient formed within the
time response of the instrument (<10 ns, Figure 3), as would
be expected for triplet formation by intersystem crossing from C-P-1C60
with a lifetime of ~1.4 ns. The decay of the transient at 690 nm was biexponential, with
lifetimes of 110 ns (85%) and 4.6 m s (15%). The lifetimes of
both components decreased when oxygen was admitted to the sample. The minor decay
component is assigned to the carotenoid triplet state, which has a small absorption in
this region. The amplitude of this component of the decay is only ~0.4% that of the
carotenoid triplet-triplet absorption at 550 nm. The major component of the decay at 690
nm is ascribed to C-P-3C60. The fact that its time constant is
identical to that found for the rise of 3C-P-C60 confirms that the
fullerene triplet is the precursor of the carotenoid triplet state, and that the
carotenoid triplet state was formed via triplet-triplet energy transfer. Based on these
results, a similar path for formation of the carotenoid triplet state is assumed for triad
1. The fact that the yield of carotenoid triplet state in both molecules is unity,
within experimental error, verifies that no other decay pathways for the porphyrin or
fullerene excited singlet manifolds can compete with singlet-singlet energy transfer from
the porphyrin to the fullerene followed by intersystem crossing in C-P-1C60.
2.3 Photochemistry at low temperatures
What is the mechanism of the
triplet-triplet transfer? Two pathways are conceivable. One of these is a single-step
transfer from the fullerene to the carotenoid (step 16 in Figure 1),
whereas the second involves an energy transfer relay such as those seen in photosynthetic
reaction centers. In the relay mechanism, endergonic energy transfer from the fullerene to
the porphyrin (step 11) is followed by rapid, exergonic transfer to the carotenoid (step
12). Excitation of the porphyrin moiety of carotenoporphyrin dyad 3 is followed by
intersystem crossing to the porphyrin triplet state, which transfers energy to the
carotenoid to form 3C-P within 10 ns. Thus, if the two-step pathway is followed
in 1 and 2, the initial step 11 must be the rate-limiting step. The
temperature dependence of the triplet energy transfer was investigated in order to help
distinguish between the two mechanistic possibilities.
Excitation of 1 in a 2-methyltetrahydrofuran glass at 77K gave
rise to several transient species (Figure 4).[25] The C· + -P-C60· - state
was detected via observation of the carotenoid radical cation absorption at 980 nm (Figure 4b, F ~
0.10). None of the transient decays at 77K are true single exponentials, probably due to
conformational or environmental heterogeneity. The decay at 980 nm can be fitted as two
exponentials with a major component (73%) having a 1.5 m s
lifetime and a minor, ~7 m s component. The 1.5 m s decay is accompanied by a corresponding rise of the carotenoid
triplet absorption at 550 nm with a major 1.4 m s component.
The 3C-P-C60 species (F ~ 0.07) decays with a lifetime of 10 m s (Figure 4c). A transient with a maximum absorbance at ~700 nm,
corresponding to C-P-3C60, forms with the excitation pulse in a
yield of 0.17 and decays with a major decay component of 81 m s
(Figure 4a).
These results show that in the glass at 77 K, C-P-3C60
is formed within 10 ns of excitation with a quantum yield of 0.17 and decays with a rate
constant of 1.2 X 104 s-1. At 590 nm, ~19% of the absorbed light
excites the fullerene directly. The yield of intersystem crossing in fullerene model
compounds is essentially unity. At 77 K, C-P-1C60 is not quenched by
electron transfer, but decays by intersystem crossing. Thus, the observed fullerene
triplet state arises mainly from light absorbed directly by the fullerene moiety. The
majority of the C-1P-C60 states decay by photoinduced electron
transfer step 3 to give C-P· + -C60· - , which goes on
to yield C· + -P-C60· - via step 4 with an overall
yield of 0.10. The final C· + -P-C60· - charge-separated
state slowly recombines to yield the carotenoid triplet (k8 + k9
= 6.7 X 105 s-1), which decays with k15 = 1.0 X 105
s-1. The triplet lifetime of a model fullerene similar to the fullerene moiety
of 1 and 2 is 89 m s at 77 K in a
2-methyltetrahydrofuran glass, whereas the lifetime of the C-P-3C60
is 81 m s under the same conditions. Thus, triplet-triplet
energy transfer in 1 does not occur at 77 K, and C-P-3C60
decays to the ground state by the usual pathways.
The sharp contrast between the decay behavior of C-P-3C60
at ambient temperature and at 77 K suggested that a more detailed study of the temperature
dependence of this decay would be fruitful. A ~1 X 10-3 M solution of 1 in
2-methyltetrahydrofuran was excited with a ~5 ns, 625-nm laser pulse and the transient
absorption kinetics at 700 nm were determined at various temperatures. Illustrative decays
at 16, 130 and 190 K are shown in Figure 5. The decays as
determined under these conditions are two exponential, with a major, long component and a
minor shorter component. In each case, the short part of the decay correlates with the
decay at the carotenoid radical cation absorption maximum in the 950 nm region. This part
of the decay results from transient absorption by the C· + -P-C60·
- charge-separated state, which decays to 3C-P-C60.
The longer component of the decay at 700 nm is assigned to the fullerene triplet state.
Thus, C-P-3C60 decays as a single exponential with time constants of
3.8 m s, 55 m s, and 70 m s at 190, 130 and 16 K, respectively. Figure
6 shows a plot of the rate constant for decay of C-P-3C60
against 1000/T for 12 temperatures. It will be noted that the decay process is
strongly activated down to about 150 K, and temperature invariant thereafter.
3. Discussion
The results presented above show that in
toluene solution, neither triad 1 nor the closely related 2 undergo
photoinduced electron transfer, although they do so in high yield in more polar solvents.
The only significant pathways for decay of both C-1P-C60 and C-P-1C60
result ultimately in the formation of 3C-P-C60 with unity quantum
yield. This occurs via the transfer of singlet excitation from the porphyrin to the
fullerene followed by normal intersystem crossing in the fullerene to yield C-P-3C60.
Triplet energy is then efficiently transferred to the carotenoid.
The remaining question concerns the mechanism of the transfer. The
possibilities are a single-step transfer by path 16 in Figure 1,
or a two-step relay involving slow transfer from the fullerene to the porphyrin via step
11 followed by rapid transfer by step 12 from the porphyrin to the carotenoid. The studies
of 1 in 2-methyltetrahydrofuran allowed direct observation of the fullerene triplet
state and its decay properties as a function of temperature. The temperature dependence
shown in Figure 6 suggests an energy transfer process with a
significant energy of activation which is effective down to about 150 K. At lower
temperatures, the decay of C-P-3C60 is activationless. The activated
process is exactly what one would expect for a triplet relay whose first step is
endergonic. By contrast, single-step, exergonic triplet energy transfer from the porphyrin
moiety to the carotenoid of a carotenoporphyrin with a structure related to 3
occurs within a few ns even at 77 K.[34]
The activation energy Ea for triplet-triplet energy
transfer may be determined from the temperature data. The solid line in Figure 6 is a fit of the rate data (kobs) to eq
1, which is
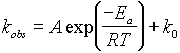 (1)
(1)
simply the Arrhenius equation with an additional rate
constant, k0, that describes the activationless portion of the decay.
The fit yields an Arrhenius activation energy of 0.17 eV, a k0 of 1.3 X
104 s-1, and a pre-exponential factor A of 9.5 X 109
s-1. This value of Ea must be a maximum for the energy
difference between the fullerene triplet and the more energetic porphyrin triplet state.
The energy of the lowest-lying triplet state of fullerene 4 is reported to be 1.50
eV.[35] The triplet energy of free base octa-alkylporphyrins
is typically ~1.6 eV.[36] This suggests that the minimum
energy of activation for the proposed triplet energy transfer relay should be about 0.1
eV, which is consistent with the measured activation energy for the transfer in 1.
These results suggest that the triplet relay does indeed operate in
triads 1 and 2 down to about 150 K. At this temperature, the activated
process becomes too slow to compete with deactivation of the fullerene triplet by other
mechanisms, presumably return to the ground state by intersystem crossing and
phosphorescence. The natural photosynthetic triplet energy transfer relay also involves an
endergonic step, and its rate is also strongly temperature dependent. The rate of transfer
slows considerably below about 50 K, and at 10 K, triplet transfer to the carotenoid is
not observed.[22,23,37,38]
4. Conclusions
These fullerene-containing triads are thus able to mimic the photophysics of the triplet energy transfer relay that has been postulated to operate in bacterial reaction centers [22-24] and possibly in PSII reaction centers.[15,16] Although model systems have been prepared in the past that demonstrate both single step[4,5,31,34,39-42] and, in one example, two-step[43] triplet-triplet energy transfer to carotenoids, the fullerene systems are the first to also demonstrate other important properties reminiscent of natural reaction centers. For example, triad 1 features singlet-singlet energy transfer from the porphyrin to the fullerene moiety (antenna function), and photoinduced multistep electron transfer to give a long-lived, energetic charge-separated state in high yield. Electron transfer occurs even at low temperatures (down to at least 10 K); this behavior is unusual in reaction center models. Finally, charge recombination of C· + -P-C60· - occurs to give the carotenoid triplet state, rather than the ground state. This behavior, too, is rare in model reaction centers.
5. Experimental section
Ultraviolet-visible spectra were measured
on a Shimadzu UV2100U UV-VIS Spectrometer, and fluorescence spectra were measured on a
SPEX Fluorolog using optically dilute samples and corrected. Fluorescence decay
measurements were performed on ~1 X 10-5 M solutions by the time-correlated
single photon counting method. The excitation source was a cavity-dumped Coherent 700 dye
laser pumped by a frequency-doubled Coherent Antares 76s Nd:YAG laser.[44] The instrument response function was 35 ps, as measured at
the excitation wavelength for each decay experiment with Ludox AS-40.
Transient absorption measurements on the picosecond
time scale were made using the pump-probe technique. The sample was dissolved in purified
solvent and the resulting solution was circulated by magnetic
stirring in a cuvette having a 2-mm path length in the beam area. Excitation was at 590 nm
with 150-200 fs, 30 m J pulses at a repetition rate of 540 Hz.
The signals from the continuum-generated white-light probe beam were collected by an
optical spectrometric multichannel analyzer with a dual diode array detector head.[45]
Nanosecond transient absorption measurements were made with
excitation from an Opotek optical parametric oscillator pumped by the third harmonic of a
Continuum Surelight Nd:YAG laser. The pulse width was ~5 ns, and the repetition rate was
10 Hz. The detection portion of the spectrometer has been described elsewhere.[46]
Acknowledgments
This work was supported by the National Science Foundation (CHE-9709272). This is publication 343 from the ASU Center for the Study of Early Events in Photosynthesis.