Concerning the Triplet Energies of Non-metallated Phthalocyanines and the Mechanism of Singlet Oxygen (1Dg) - sensitized Phthalocyanine Luminescence
D. M. Baigel, A. A. Gorman,* I. Hamblett and T. J. Hill
Department of Chemistry, University of Manchester, Manchester M13 9PL, U. K.
Abstract: Establishment of equilibria for triplet energy transfer between singlet oxygen (1Dg), and both 2,3,9,10,16,17, 23,24-octahexadecyl- phthalocyanine (OHPC) and tetra-t-butylphthalocyanine (TBPC)
(a) demonstrates that the identical triplet energies of these non-metallated phthalocyanines, 22.8 ± 0.2 kcal mol-1, are very significantly less than hitherto thought and (b) provides strong support for a mechanism of 1Dg - sensitized phthalocyanine luminescence which involves triplet energy transfer fom 1Dg to the phthalocyanine triplet, these two species being the electronically excited components of a triplet energy transfer equilibrium.
1. Introduction
The triplet energy of the basic phthalocyanine chromophore is a parameter of central importance to the controversy concerning the mechanism of the
1Dg -sensitized luminescence of phthalocyanines1-4. Whereas metallo-phthalocyanines phosphoresce, this is not the case for the parent chromophore5 and the only reported triplet energy for the latter is that of 28.7 kcal mol-1 6 determined from a Sandros treatment of triplet energy transfer rate constants. Although data points in Ref. 6 are sparse and fail to demonstrate what must in reality be classical behaviour, it is apparent to us that an isoenergetic tripet energy would be much closer to that for b-carotene, known to have a vertical triplet energy of 23.0 ± 0.3 kcal mol-1.7 In this work we describe accurate determination of the triplet energy of 2,3,9,10,16,17,23,24- octahexadecylphthalocyanine (OHPC) used in our sensitized luminescence work.3 Using the same technique we have obtained exactly the same triplet energy for tetra-t-butyl-phthalocyanine (TBPC) employed by Krasnovsky and Foote.2,4 The significance of this value is discussed.
2. Experimental
2.1 Time-resolved Experiments Pulse radiolysis experiments were essentially as described.8 Laser flash experiments utilised the 12 ns, 355 nm third harmonic of a
J. K. Lasers 2000 Q-switched Nd-YAG laser. The 1270 nm luminescence detection system9 and the triplet-triplet absorption experiments10 were essentially as previously described. Digitized data were processed on an MJN386SX personal computer. All pulsed experiments were single shot on fresh solutions.
2.2 Materials OHPC,11 and benzene12 were as described. Benzene-d6 (Sigma, 99.5%) and TBPC (Aldrich, 99%) were used as received.
3. Results and Discussian
The triplet, 3OHPC*, was initially characterised in deaerated benzene using a combination of standard pulse radiolysis and laser flash photolysis techniques to be fully described elsewhere. Parameters determined were lmax 500 nm (e500 = 32,500 ± 5,000 l mol-1 cm-1), fT = 0.19 ± 0.04, tT = 143 ± 23 ms and kT = (7.0 ± 1.0) x 103 s-1. The decay of the triplet was absolutely clean, i.e. no process other than decay to ground state was observed.
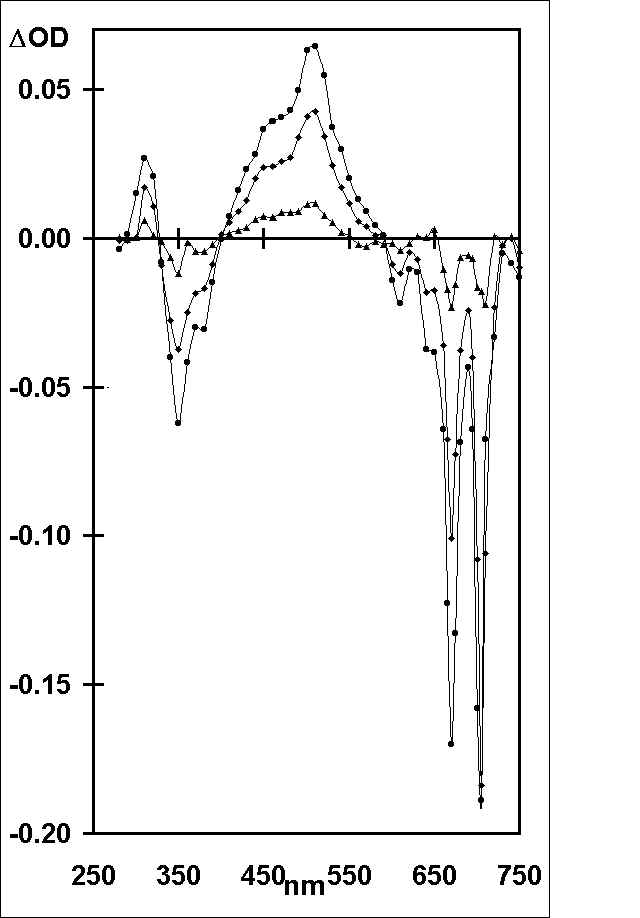
In Figure 1 are shown transient absorption/bleaching spectra following laser excitation (355 nm; 15 ns pulse) of OHPC (1.05 x 10-5 mol l-1; A355 = 0.63) in benzene containing a low concentration of oxygen (2.2 x 10-4 mol l-1). The spectra which are a combination of the triplet absorption and corresponding ground-state bleaching of OHPC are identical to those observed by pulse radiolysis and essentially unchanged over the whole time profile. The kinetics of the transient absorption/bleaching decay were (a) independent of wavelength and (b) biexponential (Figure 2(a)), showing rapid decay to an equilibrium followed by slow bleeding of that equilibrium (cf. eq 1). Extrapolation of the fast and slow
 click
for full size figure
click
for full size figure
Figure 1. Transient absorption/bleaching changes with time after laser excitation of OHPC (A355 = 0.63) at 355 nm in benzene containing oxygen (2.2 x
10-4 mol l-1), measured at 2.7 (circles), 5.0 (diamonds) and 25 (triangles) ms, after the laser pulse.
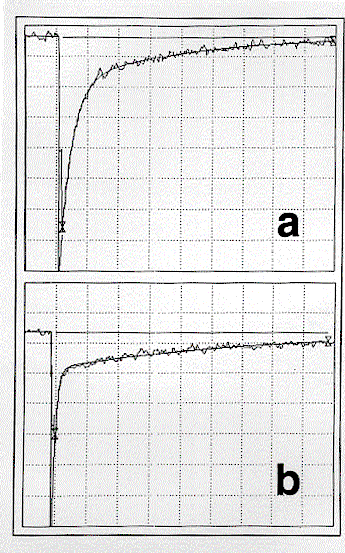
Figure 2. (a) Transient absorption corresponding to Figure 1, at 500 nm, 2.1% absorption/division, 10 ms/division, k1 = 2.8 x 105 s-1(calc. 3.2 x 105 s-1), k2 = 2.8 x 104 s-1 (calc. 2.7 x 104 s-1), Ai = 7.4 x 10-2, Ae = 1.7 x 10-2. (b) As for (a) with A355 = 0.57 in benzene-d6, 0.96% absorption/division, 50 ms/division, k1 = 2.6 x 105 s-1 (calc. 3.2 x 105 s-1), k2 = 3.1 x 103 s-1 (calc. 2.8 x 103 s-1), Ai = 2.2 x 10-2, Ae = 4.7 x 10-3.
components of the triplet decay to time zero (cf . Figure 2(a)) gave respectively the absorbances of 3OHPC* both initially, Ai, and at establishment of equilibrium , Ae.
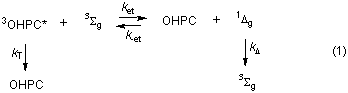
The equilibrium of eq 1, defined as K = [Ai - Ae] [OHPC] [Ae]-1 [3Sg]-1 = 0.17 ± 0.04, is displaced to the left by the spin statistical factor of 1/9 associated with ket.13 Appropriate correction and a standard assumption of no entropic changes gave DH = -0.25 ± 0.16 kcal mol-1 and a value of 22.8 ± 0.2 kcal 1Dg for the triplet energy of OHPC. This is very significantly different to the published figure of 28.7 kcal mol-1 for phthalocyanine itself 6 and, as anticipated, is very close to that of b-carotene.
Determination of ket as 1.2 x 109 l mol-1 s-1 in experiments using much higher oxygen concentrations allowed a value of 7.1 x 109 mol l-1 to be assigned to k-et. Our values for ket, k-et, and kT, together with kD = 3.2 x 104 s-1 14 (cf. eq 1) allowed calculation13,15 of the first-order constants for decay to (k1) and decay of (k2) the equilibrium of eq 1 which were in good agreement with experiment (cf. Figure 2(a)).
Laser excitation in deaerated benzene-d6 resulted in the same clean formation and decay of 3OHPC* with an identical kT. However, whereas in the presence of 2.2 x 10- 4 oxygen (cf. Figure 2(b)), decay to equilibrium proceeded with an essentialy identical k1, the rate constant for decay, k2, of that equilibrium was much slower, 3.1 x 103 s-1 since kD is now only 1.7 x 103 s-1.14 The calculated13,15 value is clearly in good agreement with experiment. Thus, in this benzene-d6 experiment, 3OHPC* decays by a factor of 2-3 slower than in the deaerated medium, further evidence for the establishment of an equilibrium with1Dg. As expected, in both benzene and benzene-d6 , the time profile of the 1270 nm emission of the 1Dg state followed that of the long-lived triplet decay. The long risetime of the emission detection system coupled with immediate phthalocyanine fluorescence prevented observation of the 1Dg emission growth.The equilibrium constant for benzene-d6, determined as described above, was identical to that in benzene as were the corresponding benzene and benzene-d6 values for TBPC.
In the published work where time-resolved 1Dg-sensitized phthalocyanine (PC) fluorescence has been observed,2-4 the equilibrium of eq 3 will have been established before monitoring of the delayed fluorescence began, i.e. the key concentrations are equilibrium concentrations. For instance, we have determined the rate constants for quenching of 2-acetonaphthone triplet by OHPC and by oxygen to be 7.5 x 109 l mol-1 s-1 and 1.6 x 109 l mol-1 s-1 respectively, essentially identical to ket and k-et respectively as one might anticipate from the nature of the processes involved. Thus, in our sensitized luminescence experiments3 the developing relative concentrations of 1Dg and 3OHPC* will be essentially those of the equilibrium situation. If the phthalocyanine absorbs any of the incident light (~ 5 % in our experiments)3 the initial 3OHPC* concentration will be higher than that at equilibrium which will then be established within 500 ns in aerated benzene.The knowledge that we are dealing with an equilibrium situation allows us to conclude that the sensitization event in the 1Dg -sensitized production of PC fluorescence is that of the second step depicted in eq 2. It is spin-allowed, exothermic by at least
![]()
4.6 kcal mol-1 and, based solely on calculated equilibrium concentrations in aerated benzene (or benzene-d6), 3000 times more probable than any mechanism
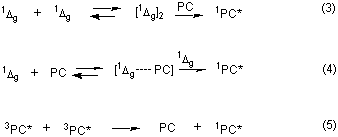
involving two 1Dg molecules and a PC ground state in the sensitization step (cf. eqs 3 and 4).2,4 It is only 500 times more probable than the classical triplet-triplet annihilation process (cf. eq 5) which is also a clear mechanistic contender. These ratios are independent of the phthalocyanine concentration employed when the oxygen concentration is orders of magnitude higher. Thus, an accurate knowledge of the phthalocyanine triplet energy provides strong support for our conclusion2 that 1Dg -sensitized phthalocyanine luminescence is a further example of the well-documented Ogryzlo-Pearson-Wilson mechanism16,17, involving clear sequential transfers of triplet energy from two 1Dg molecules, first to the PC ground state, secondly to the thus formed PC triplet state.
We wish to thank the University of Manchester Samuel Gratrix Fund and the EPSRC (U.K.) for financial support and Dr. N. B. McKeown for a generous gift of OHPC. The experiments described were performed at the Paterson Institute for Cancer Research Free Radical Research Facility, the Christie Hospital NHS Trust, Manchester. The Facility is funded under the European Commission TMR PROGRAMME - ACCESS TO LARGE SCALE FACILITIES, Grant ERBFMGECT 950084 - Access to PICR FRR Facility.
References
(1) A. A. Krasnovsky Jr.and K. V. Neverov, Biofizica (Sov. Biophys.) 1988, 23, 884. A. A. Krasnovsky Jr.and K. V. Neverov, Chem. Phys. Lett. 1990, 167, 591. K. V. Neverov and A. A. Krasnovsky Jr., Opt. Spektrosk. (Sov. Opt. Spectrosc.) 1991, 71, 691.
(2) A. A. Krasnovsky, Jr. and C. S. Foote, J. Am. Chem. Soc. 1993, 115, 6013.
(3) A. A. Gorman, I. Hamblett and T. J. Hill, J. Am. Chem. Soc., 1995, 117, 10751.
(4) S. Murphy and C. S. Foote, Photochem. Photobiol. 1997, 65(S), 1S.
(5) P. S. Vincett, E. M. Voigt and K. E. Rieckhoff, J. Chem. Phys. , 1971, 55, 4131.
(6) J. McVie, R. S. Sinclair and T. G. Truscott, J. Chem. Soc., Faraday Trans. II , 1978, 74, 1870.
(7) A. A. Gorman, I. Hamblett, C. Lambert, B. Spencer and M. C. Standen, J. Am. Chem. Soc. , 1988, 110, 8053.
(8) A. A. Gorman and I. Hamblett, Chem. Phys. Lett. 1983, 97, 422.
(9) A. A. Gorman, A. A, I. Hamblett, C. Lambert, A. L. Prescott, M. A. J. Rodgers and H. M. Spence, J. Am. Chem. Soc. 1987, 109, 3091.
(10) A. A. Gorman, I. Hamblett, M. Irvine, P. Raby, M. C. Standen and S. Yeates,
J. Am. Chem. Soc. 1985, 107, 4404.
(11) G. J. Clarkson, N. B. McKeown and K. E. Treacher, J. Chem. Soc., Perkin Trans. I 1995, 1817.
(12) A. J. G. Barwise, A. A. Gorman, R. B. Leyland, P. G. Smith and M. A. J. Rodgers, J. Am. Chem. Soc. 1978, 100, 1814.
(13) P. A. Firey, W. E. Ford, J. R. Sounik, M. E. Kenney and M. A. J. Rodgers, J. Am. Chem. Soc. , 1988, 110, 7626.
(14) A. A. Gorman and M. A. J. Rodgers, Handbook of Organic Photochemistry ,
Vol. 1, ed. J. C. Scaiano, CRC Press, Boca Raton, 1989, pp. 229-247.
(15) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, New York, 1970, pp. 301-311.
(16) E. A. Ogryzlo and A. E. Pearson, J. Phys. Chem. , 1968, 72, 2913.
(17) T. Wilson, J. Am. Chem. Soc., 1969, 91, 2387.