GROUND AND EXCITED PROPERTIES OF THE PREVITAMIN D CONFORMERS
Olga Dmitrenko
Institute of Surface Chemistry, National Academy of Sciences of Ukraine,
252022 Kiev, Ukraine
Wolfgang Reischl
Institute of Organic Chemistry,
University of Vienna, A-1090 Vienna, Austria
James T. Vivian and John H. Frederick
Department of Chemistry, University of
Nevada, Reno, NV 89557, USA
Abstract. Ground and excited states properties of previtamin D
individual conformers are examined by use of the MMX (PCMODEL) empirical force field, and
AM1 and QCFF/pi semi-empirical calculations. Ground state geometries obtained by these
methods indicate a highly twisted triene system with similar degrees of non-planarityfor
all conformers. The results for origin transitions support the suggestion that cZc
conformers absorb at longer wavelength than tZc ones, thereby contributing to the
understanding of wavelength dependence of previtamin D photochemistry.
Keywords. Previtamin D conformers, ground and excited states, force-field
calculations,
AM1 conformational analysis, QCFF/PI calculations, absorption properties of conformers
1.Introduction
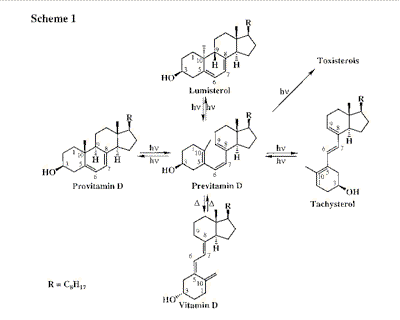
The photochemical reaction of previtamin D formation from the steroidal diene precursor,
provitamin D (7-dehydrocholesterol, Scheme 1), occurs in mammal skin and is the key step
in the biogenesis of vitamin D [1]. The mechanisms of the photoreactions involved in
previtamin D photosynthesis are still the subject of intensive investigations [2,3].
Reaction scheme of previtamin D photosynthesis (In vivo the side-reactions,
cis-trans isomerization into tachysterol and irreversable photoconversions into
toxisterols, are inefficient).
One of the open questions in previtamin D photochemistry is its wavelength dependence
[3-5]. To gain a better understanding of this dependence, a detailed knowledge of the
ground and excited states of the various conformers of this flexible steroid molecule is
necessary. It has been suggested [3 and references therein] that the origin of the sudden
change of quantum yields of previtamin D photoconversions at the red edge of the
absorption band can be attributed to the selective excitation of ground state conformers
that differ in their electronic absorption characteristics. Up to now little is known
about the conformational population of previtamin D and, moreover, about the absorption
properties of its individual conformers [3].
Here we will examine the previtamin D ground state conformations and the associated
transitions to the lowest "bright" singlet excited state. The aim of this work
is to calculate the absorption properties of previtamin D conformations and to show how
they might contribute to the wavelength dependence of the photoreaction.
2. Computational details
For the ground-state conformational analysis we use the MMX force field from PCMODEL
[6] and AM1 semi-empirical calculations [7]. In all the previtamin D structures, the
hydrocarbon side chain has been substituted by a methyl group for simplicity. The first
singlet excited states of 3-desoxy-previtamin D conformers(1)
have been studied by the QCFF/pi program [8] developed by A.Warshel and M.Karplus for the
calculation of the ground and excited states of large conjugated molecules. Like MMX,
QCFF/pi is based on the formal s -- p
separability: the F-potential surface is represented by empirical potential functions
whereas B-electrons are treated by a semiempirical approach (Pariser-Parr-Pople type). In
QCFF/pi, only singly excited B-electron configurations are included in the CI matrix. A
number of studies (e.g. on stilbenes [9]) indicate that QCFF/pi computations are suitable
for cases of large conformational changes, thereby supporting our choice for modeling the
steroid trienes which are highly conformationally flexible and strongly distorted by
steric interactions. The starting geometries of 3-desoxy-previtamin D conformers have been
taken from the previous MMX optimizations of previtamin D by use of the default
(charge-charge) calculation scheme of intramolecular electrostatic interactions [10].
3. Results and discussion
The detailed molecular-mechanics-based conformational search on previtamin D molecule and its analogs have been presented earlier in [10]. The results obtained there support the prognostic quality of MMX conformational analysis. However our recent computational studies [11] motivated by ultrafast studies of Fuss et al. [12] have revealed the serious problem in modeling a highly flexible molecule possessing a polar OH-group, namely, that the relative stabilities of different conformers are very sensitive to the modeling approach and, in particular, to the intramolecular interactions calculation scheme [11].
In contrast to the relative stabilities, the different previtamin D conformations
themselves are somewhat reproducible using different computational approaches as is
demonstrated in Table 1. For the description of individual conformers the following
abbreviations are used: Z denotes the cis geometry of the C6=C7 double bound; letters c
and t refer to the cis and trans geometries of C5-C6 and C7-C8 single bonds respectively;
(-) or (+) indicates the sign of these torsion angles according to the rules of Klyne and
Prelog [13].
Table 1. Selected torsion angles of previtamin D ground state conformers optimized by MMX- and AM1 semi-empirics.
Conformer |
C10=C5-C6=C7 |
C6=C7-C8=C9 |
||
MMX |
AM1 |
MMX |
AM1 |
|
OH-equatorial |
||||
| (-)cZ(-)c | -70 |
-80 |
-43 |
-32 |
| (+)cZ(+)c | 57 |
78 |
41 |
29 |
| (-)tZ(+)c | -123 |
-121 |
43 |
29 |
| (+)tZ(-)c | 149 |
121 |
-45 |
-30 |
| (+)tZ(+)t | 133 |
122 |
125 |
104 |
| (-)cZ(+)t | -51 |
- |
132 |
- |
OH-axial |
||||
| (-)cZ(-)c | -66 |
-81 |
-43 |
-22 |
| (+)cZ(+)c | 61 |
87 |
45 |
24 |
| (-)tZ(+)c | -147 |
-117 |
54 |
29 |
| (+)tZ(-)c | 116 |
116 |
-38 |
-26 |
| (+)tZ(+)t | 136 |
120 |
134 |
116 |
| (-)cZ(+)t | -47 |
- |
133 |
- |
The occurrence of conformers with large values of these torsion angles (independently derived from the different calculation approaches) allows us to conclude that these highly twisted non-planar structures are realistic and contribute significantly to the conformer population. This justifies their use as input data for UV spectra calculations. We believe that all twelve conformations found in our earlier work [10] contribute to UV absorption, thereby accounting for the absence of any structure in the absorption spectrum of previtamin D (see Figure) even at low temperature[4,5]
.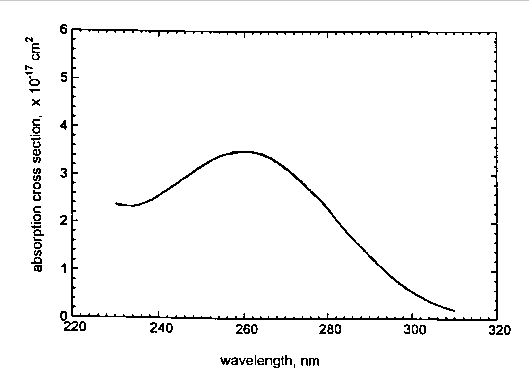
UV absorption spectrum of previtamin D
Since the QCFF/pi program does not provide parameters for a hydroxyl group, the 3b-OH group in previtamin D was replaced by a hydrogen atom.
We used the MMX derived previtamin D geometries as input structures for the QCFF/pi calculations and due to the half-chair -- half-chair interconversion of the A-ring, again two sets of conformers have been obtained. In one set of six conformers, the 3�-hydrogen is in equatorial orientation; whereas, in the other six conformers the 3�-hydrogen is found in an axial orientation. Selected data for the ground- and first allowed excited state geometries of 3-desoxy-previtamin D conformers are given in Table 2. Comparison of the ground state torsion angles obtained after minimization by QCFF/pi with initial (MMX optimized) angles reveals no significant changes (Table 1 and Table 2). Thus, re-optimization by QCFF/pi does not cause any marked changes in the ground state geometries of the steroid triene system.
Some trends are observed for the geometry changes in the conformers upon
excitation. All the optimized excited singlet conformers possess more planar torsion
angles about the single bonds and significantly distorted planarity (up to 45deg) about
the central doble bond, C5-C6=C7-C8.
Table 2. Changes in the torsion angles of triene system of desoxy-previtamin D conformers upon excitation. The ground state values are given in parentheses.
| Conformer | C10=C5-C6=C7 | C5-C6=C7-C8 | C6=C7-C8=C9 |
3�-H equatorial |
|||
| (-)cZ(-)c | -33 (-68) | -42 (-10) | -19 (-43) |
| (+)cZ(+)c | 23 (52) | 35 (7) | 16 (37) |
| (-)tZ(+)c | -150 (-126) | 40 (9) | 16 (39) |
| (+)tZ(-)c | 173 (151) | -45 (-14) | -21 (-45) |
| (+)tZ(+)t | 161 (133) | -43 (-11) | 144 (121) |
| (-)cZ(+)t | -24 (-50) | -37 (-6) | 150 (129) |
3�-H axial |
|||
| (-)cZ(-)c | -30 (-62) | -41 (-12) | -17 (-39) |
| (+)cZ(+)c | 26 (57) | 36 (7) | 18 (40) |
| (-)tZ(+)c | -170 (-147) | 42 (11) | 26 (52) |
| (+)tZ(-)c | 145 (121) | -43 (-11) | -14 (-36) |
| (+)tZ(+)t | 161 (136) | -41 (-9) | 152 (130) |
| (-)cZ(+)t | -21 (-46) | -37 (-6) | 151 (130) |
The characteristic data upon excitation of the previtamin D conformers are summarized in Table 3. Vertical transitions (which reflect the absorption maximum) do not appear to be specifically dependent on the triene geometry; whereas, the values of the oscillator strength clearly suggest that conformers with at least one s-trans bond possess a stronger absorption [1]. The results for the origin transitions clearly show that cZc conformations absorb at longer wavelength than those containing at least one s-trans bond. This is in agreement with earlier predictions suggested in numerous studies (see, for example [1]) as well as with the recent measurements of Fuss and Lochbrunner [5]. They have measured the low temperature UV spectrum of cZc conformer just formed from its precursor, provitamin D, and compared it with the low temperature spectrum for equilibrated previtamin D. At wavelengths longer than 310 nm, the absorption of cZc conformer was far larger than the absorption of tZc [5].
Table 3. Electronic transitions of desoxy-previtamin D conformers*.
| Conformer | Excitation energy, cm-1 (wavelength, nm) | Oscillator strength | wavelength 0-0, nm |
3�-H equatorial |
|||
| (-)cZ(-)c | 40343.1 (248) | 0.35 | 314 |
| (+)cZ(+)c | 37134.0 (269) | 0.3 | 325 |
| (-)tZ(+)c | 38857.1 (257) | 0.39 | 307 |
| (+)tZ(-)c | 37310.9 (268) | 0.58 | 311 |
| (+)tZ(+)t | 42074.7 (238) | 0.73 | 283 |
| (-)cZ(+)t | 39213.9 (255) | 0.45 | 301 |
3�-H axial |
|||
| (-)cZ(-)c | 38626.6 (259) | 0.35 | 318 |
| (+)cZ(+)c | 38274.0 (261) | 0.3 | 323 |
| (-)tZ(+)c | 38795.9 (258) | 0.52 | 304 |
| (+)tZ(-)c | 38956.9 (257) | 0.38 | 307 |
| (+)tZ(+)t | 40637.7 (246) | 0.85 | 284 |
| (-)cZ(+)t | 38571.2 (259) | 0.47 | 304 |
*In the results presented, only singly excited B-electron
configurations were included in the CI matrix. Thus, just the allowed 1B2 singlet excited
state mostly contributing to the UV absorption is under consideration [14].
Thus, one may attribute the different photochemical behaviour at the red
edge of previtamin D absorption band to the selective excitation of cZc conformers. This
also may be one of the reasons for the dramatic increase in the efficiency of toxisterols
formation observed by Terenetskaya et al. [15] upon long-wavelength photolysis of
provitamin D. However, Fuss and Lochbrunner have recently postulated [5] that the dominant
origin of wavelength dependence in previtamin D photochemistry may be caused by an
isomerization process (double bond isomerization) on the excited state surface which
possesses an energy barrier. As the excitation wavelength is shortened this process
compete with other barrierless reactions, thereby altering all the quantum yields[5].
4.Conclusions
Our results, obtained using a variety of different computational methods, indicate a great predominance of non-planar geometries for the individual previtamin D conformers than had been found earlier by MMP2 computational studies alone [16]. Thus, the highly twisted triene moiety of previtamin D molecule (which plays a central role in its structural and photophysical properties) must be properly accounted for when extrapolating from the properties of simpler model trienes.
In addition our results for the origin excitations support the earlier
assumptions of Jacobs and Havinga [1], namely, that cZc conformers absorb at longer
wavelengths than tZc ones. This means that it is important to consider both the excited
state effects [5] and selective excitation of ground state conformations [3], when
discussing the wavelength dependence of previtamin D photosynthesis. The present results
are performed for gas-phase conformations and reflect a general tendency. For qualitative
comparison with the experimental observations they require futher corrections for solvent
effects. Moreover, for non-planar structures the s -- p separation is no longer strictly valid; thus, the results obtained
here should be considered as a qualitative first approximation.
5. Acknowledgments
We are grateful to W. Fuss for valuable e-mail discussions. O.D.
acknowledges financial support through the U.S. National Science Foundation (CHE-9419102
to J.H.F.) and a Fellowship from the President of Ukraine. Access to an SGI workstation
was made possible by a grant of the Fonds z�r F�rderung der wissenschaftlichen Forschung
(P09859-CHE, to W. Weissensteiner). Financial support from the NSF and �sterreichische
Nationalbank (Jubilaeumsfondsprojekt Nr.: 4865) are gratefully acknowledged.
6. References
| 1 | H.J.C.Jacobs and E.Havinga, Adv. Photochem. 11 (1979) 305-373; A.W. Norman, "Vitamin D, The Calcium Homeostatic Steroid Hormone" Academic Press: New York, 1979 |
| 2 | F.Bernardi, M.Olivucci, I.N.Ragazos and M.A.Robb, J.Am.Chem.Soc. 114 (1992) 8220-8225 |
| 3 | H.J.C.Jacobs, Pure Appl.Chem.67 (1995) 63-70; A.M.Brouwer, J. Cornelisse, H.J.C.Jacobs,Tetrahedron 43 (1987) 435-438 |
| 4 | W.G. Dauben, B.Disanyaka, D.J.H.Funhoff,B.E. Kohler, D.E. Schilke, B. Zhou, J.Am.Chem.Soc. 113 (1991) 8367-8374 |
| 5 | W. Fuss and S. Lochbrunner, J.Photochem. Photobiol.A:Chem 105 (1997) 159-164 |
| 6 | N.L.Allinger and H.L.Flanagan, J. Comput. Chem., 4 (1983), 399; G -MMX and PCMODEL (Serena Software, Bloomington, Indiana) for SGI Indigo 2. |
| 7 | M.J.S. Dewar, E.G. Zoebisch, E.F. Healy and J.J.P. Stewart, J.Am.Chem.Soc. 107 (1985) 3902-3909 |
| 8 | A.Warshel and M.Karplus, J.Am.Chem.Soc. 94 (1972) 5612-5625; Quantum Chemistry Program Exchange,Indiana University. Program No. 534 |
| 9 | A.Warshel, J. Chem. Phys. 62 (1975) 214-221 |
| 10 | O.Dmitrenko and W.Reischl, Monatsh. f�r Chemie 127 (1996) 445 -453; O. G. Dmitrenko, I.P. Terenetskaya and W. Reisch, J.Photochem. Photobiol.A: Chem. 104 (1997) 113-117 ; O. Dmitrenko and W. Reischl, Res. Chem. Intermed. (1997) in press |
| 11 | O.G.Dmitrenko and W.Reischl, J.Mol.Struc.(Theochem), accepted for publication |
| 12 | W. Fuss, T. Hoffer, P. Hering, L.P. Kompa, S. Lochbrunner, T. Schikarski
and W.E. Schmid, J. Phys. Chem. 100 (1996) 921-927; |
| 13 | W.Klyne and V.Prelog, Exerientia 16 (1960) 521-530 |
| 14 | T. Liljefors and N.L.Allinger, J.Am.Chem.Soc. 98 (1976) 2745-2749 |
| 15 | I.P.Terenetskaya, S.I.Gundorov, V.I.Kravchenko, I.K.Berik, Sov.J.Quantum. Electron. 18 (1988) 1323-11328 |
| 16 | W.G.Dauben and D.J.H.Funhoff, J.Org.Chem.53 (1988)5070-5075 |
1. QCFF/pi in the version used here is developed for conjugated hydrocarbons and, unfortunately, does not contain parameters for treatment of hydroxyl group in previtamin D molecule. Nevertheless, we do believe that the use of its desoxy- analog provides a good first approximation for the excitation energies.