Fullerenes;
Electron Accepting Building Blocks in Photoactive Donor-Bridge-Acceptor Dyads
Dirk M. Guldi
Radiation Laboratory
University of Notre Dame
Notre Dame, IN 46556
guldi@marconi.rad.nd.edu
ABSTRACT
The redox properties of pristine fullerenes and monofunctionalized fullerene derivatives in their ground and excited states are described. These compounds have drawn much attention in the last few years for the design of devices able to perform complex functions, as in molecular switches, receptors, photoconductors and photoactive dyads. These applications are generally based on the excellent electron accepting properties of the fullerene moiety. This, together with the low reorganization energy, makes C
60 and its derivatives good candidates for building blocks of more complex systems for the conversion of light into electricity or fuels. The concept of linking fullerenes to a number of interesting electro- or photoactive species and the subsequent study of intramolecular transfer dynamics between the two moieties will be elucidated as a function of excited state energy of the antenna molecule, donor-acceptor distance, and solvent polarity.The pioneering work by Kroto et al. and Krätschmer et al. regarding the initial discovery of [60]fullerene and the development of a large-scale method for their preparation, respectively, have prompted a lively interest to probe these 3-dimensional carbon allotropes in solution and to relate their properties to conventional 2-dimensional
p-systems. Furthermore, a number of solid state properties have been reported ranging from superconductivity to nanostructered devices. The spherical symmetry of C60, arranging the 60 carbon atoms in pentagon and hexagon architectures, leads to a substantial curvature of the carbon network and, in turn, to a strong pyramidalization of the individual carbon atoms with an average hybridization of sp2.278. The sum of these effects exerts a strong impact on the reactivity of the fullerene core relative to 2-dimensional carbon networks, leading, in essence, to an extremely reactive exterior but, in contrast, to a nearly inert interior.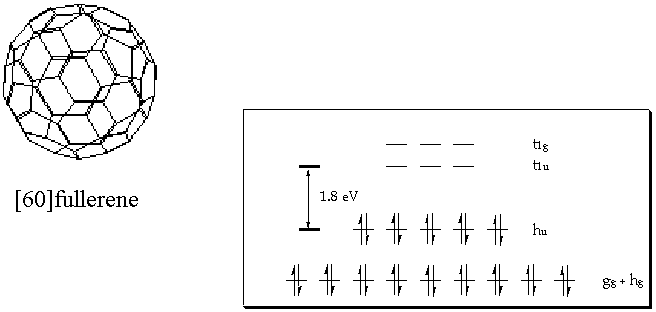
Figure 1: Structure of [60]fullerene and illustration of the pp orbital energy levels in C
608A particular intriguing consequence, which stems from this unique structure, is the electronic configuration of pristine fullerenes, comprising a five-fold degenerate HOMO (h
u) and a triply degenerate LUMO level (t1u), linked by a moderate energy gap of ca. 1.8 eV. In accord with the degeneration of the LUMO level, the redox chemistry, conducted primarily by cyclic voltammetry, demonstrated unequivocally the ability of fullerenes to accept up to six electrons in reversible one-electron reduction steps.C
60 —> (C60•-) —> (C602-) —> (C603-)—
> (C604-) —> (C605-) —> (C606-) (1)This suggests using C
60 and, to the same extent, of functionalized derivatives as potent electron acceptor moieties. The remarkably low redox potential of -440 mV versus SCE for the first one-electron reduction step, further underlines this aspect. More importantly, the exceptionally low reorganization energy of the fullerene core for such reductions is directly associated with a high degree of charge delocalization within the 3-dimensional carbon framework. Thus, forward electron transfer to the fullerene moiety is strongly favored, while, at the same time, back electron transfer experiences a noticeable slow down relative to conventionally probed electron acceptors, such as 2-dimensional aromatic molecules.A more practical aspect of fullerenes is concerned with the optical absorption spectrum of, for example, the (C
60•-) p-radical anion. This species shows a narrow band in the near IR region around 1080 nm which serves as a diagnostic probe in assisting the identification of (C60•_) and, furthermore, allows an accurate analysis of inter- and intramolecular ET dynamics in C60 containing systems.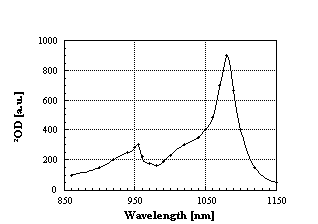
Figure 2: NIR spectrum of
the (C60•-) p-radical anion, obtained upon pulse radiolytic reduction.
Another advantage of these 3-dimensional
p-systems implements the flexibility of substituting C60 by a higher fullerene, e.g., C70, C76, C78, C84 etc. Specifically, increasing the number of carbons in the fullerene network, from C60 to C84, eases the abstraction of an electron from the corresponding HOMO level which parallels an incremental decrease of the ground state reduction potential. In other words, higher fullerenes are unequivocally even better electron acceptors and electron donors than C60.With the advent of various protocols, regarding the controlled functionalization of the fullerene core, an extensive number of functionalized fullerene derivatives became available. These chemical modifications, in fact, provide the opportunity to combine the spectacular properties of fullerenes with those of other interesting materials, including electro- and photoactive species.
The most intriguing examples, that stem from these versatile methodologies, are multicomponent donor-bridge-acceptor arrays as promising new materials for energy conversion devices. The focus of the current review is to summarize the intriguing properties of fullerenes as a multifunctional electron storage moiety. The reader should, nevertheless, be directed to a series of excellent review articles which appeared during recent years.1. Pristine [60] fullerene
Based on the symmetry prohibitions, solutions of [60]fullerene (e.g., in cyclohexane) strongly absorb in the UV region, with maxima at 260 and 330 nm that have extinction coefficients (
e) on the order of 105 M-1cm-1. In contrast, the visible region is characterized by only relatively weak transitions. For example, the maximum, noticed at 536 nm, displays an extinction coefficient of only 710 M-1cm-1. Similar to the fullerene ground state the photophysical properties of the corresponding excited states are governed by the fullerene’s 3-dimensional symmetry. A very low emission quantum yield (F) of 1.0 x 10-4, associated with the singlet excited state, relates to the combination of a short life-time, a quantitative intersystem crossing and, finally, the symmetry forbidden nature of the lowest-energy transition. To the same extent, the phosphorescence quantum yield (F < 10-6) is strongly impacted by the spherical structure of C60.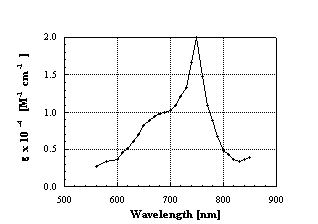
Figure 3: Triplet-triplet absorption spectrum of C
60 in toluene, obtained upon flash photolysis.In terms of transient absorption, the singlet excited state gives rise to a characteristic singlet-singlet absorption around 920 nm, whose lowest vibronic state has an energy of 1.99 eV.
, Once generated, this state is subject to a rapid and quantitative intersystem crossing process to the energetically lower lying triplet excited state (1.57 eV). In the case of [60]fullerene, this intersystem crossing takes place with a life-time of 1.3 ns, governed by a large spin-orbit coupling, which makes this process much faster than those noticed in 2-dimensional aromatic hydrocarbons. However, not only the rate is remarkable, but also the overall efficiency of the process is impressive, affording a triplet quantum yield close to unity.The triplet-triplet absorption spectrum reveals a maximum in the visible region (750 nm;
e = 20 000 M-1cm-1), and another transition in the UV (330 nm; e = 40 000 M-1cm-1). These features are easily detectable probes for uni- and bimolecular transfer reactions. The triplet life-times are dominated by triplet-triplet annihilation and ground-state quenching, and are quite short (50 and 100 ms). This is, again, much shorter than the triplet life-time of planar aromatic hydrocarbons.1.1 Bimolecular Transfer Reactions
The general processes involved in the excitation and deactivation of fullerenes in their singlet ground state and singlet / triplet excited state can be summarized as follows. Irradiation into the rich absorption features of fullerenes, which lie predominantly in the UV and lower VIS region, leads, after a series of cascade-like relaxation processes, to the formation of the singlet excited state before a rapid intersystem crossing to the triplet excited state takes place. The latter is, in turn, the starting point for a slow regeneration of the singlet ground state.
C
60 —hn—> (1C60) —ISC—> (3C60) (2)Lifting an electron from a low-energy bonding orbital to a high-energy anti-bonding orbital increases the electron affinity and, at the same time, reduces the ionization potential of the fullerene by precisely the amount of the excited state energy. Consequently, the fullerene singlet and triplet excited states are stronger oxidants and reductants than the singlet ground state. Important values, concerning the redox potentials, together with some ground state and excited state properties, of C
60, C70, and a monofunctionalized C60 derivative, are listed in Table 1. Both excited states, namely, singlet and triplet excited states, are subject to bi- and unimolecular charge transfer reactions with adequate electron acceptor and electron donor moieties. A bimolecular reaction with the initially formed singlet excited state stands, however, in direct competition with the rapidly occurring intersystem crossing and is, in essence, overshadowed by the latter. Thus, it should be noted up front that truly bimolecular transfer reactions, as outlined in the following chapters, occur with the much longer-lived triplet excited state. Alternatively, a strong interaction between the fullerene and a donor, in form of a ground state complex or excited state complex (exciplex), is also an appropriate manner to accelerate transfer dynamics evolving from the singlet excited state.A bimolecular electron transfer reaction is thought to lead to the formation of a primary contact radical pair. In the current context, this is the one-electron reduced (C
60•-) p-radical anion and the oxidized form of the applied quencher (Q•+). Specifically, the diagnostic peak of the (C60•-) p-radical anion in the near-IR region is employed as a sensitive marker whose detection points unmistakably to the generation of a charge separated radical pair, i.e., {(C60•-) / Q•+}. The latter evolves from the following sequence: an initial photoexcitation process, succeeded by a bimolecular electron transfer and, finally, a diffusional break-up of the contact pair. This dissociation depends on the ability of the environment, e.g., solvent, to retard the exergonic back electron transfer and stabilize the radical pair, for example, via strong dipole-dipole interactions.The low reduction potential of (
3C60) (+1.01 V) permits an efficient reductive quenching with a large number of amines, e.g., diazabicyclooctane (DABCO). It is noteworthy to point out that the latter does not add to ground state C60 which constitutes a fundamental advantage over primary, secondary, and tertiary amines. Selection of a polar solvent, such as alcohols or benzonitrile, is crucial in view of promoting an efficient charge separation which is, under these circumstances, typically confirmed through formation of the characteristic (C60•-) p-radical anion absorption at 1080 nm. In contrast, the low dielectric constant of toluene or chlorinated hydrocarbons prevents this charge separation process and, in turn, no appreciable life-time of the (C60•-) and (DABCO•+) radical pair is observed.(
3C60) + DABCO —> (C60•-) + (DABCO•+) (3)Similarly, ferrocene (FC), tetrathiafulvalene (TTF), bis(ethylenedithio)-tetrathiafulvalene (BEDT-TTF), 3,3’,5,5’-tetramethybenzidine (NTMB), stable nitroxide radicals, N,N-dimethyl-1-naphthylamine, polysilane, oligothiophene, and tetrathiophene moieties, to name a few representative cases, are all excellent electron donors. These compounds have therefore been probed them as sacrificial electron donors in reductive quenching reactions with triplet excited fullerenes. Again, polar solutions play a central role in creating conditions that supports an efficient charge separation and gives rise to bimolecular rate constants for the charge recombination on the order of 10
10 M-1s-1.Based on the mildly electrophilic character of C
60, formation of fullerene charge transfer complexes is not only limited to NTME. In fact, aromatic amines, such as N,N-dimethylaniline (DMA), N,N-diethylaniline (DEA), diphenylamines (DPA), triphenylamine (TPA), triethylamine (TEA), and a number of substituted naphthalenes give rise to similar behavior. Generally, the resulting complexes are quite weak, with formation constants on the order of 0.1. Accordingly, high amine concentrations are required to study the characteristics and properties of these fullerene containing charge-transfer complexes. At low amine concentrations the fullerene excited states are, however, effectively quenched through electron transfer interactions. In aliphatic hydrocarbon solvents such as hexane or methylcyclohexane, the quenching of the monomer excited states occurs via the transient formation of exciplexes. The associated exciplex emissions are extremely sensitive to the solvent environment which in polarizable benzene and toluene, for example, leads to a complete quenching of the exciplex.Finally, a bimolecular electron transfer with a ground state zinc phthalocyanine (ZnPc) exhibits an interesting twist. The oxidation potential of this chromophore, generating the ZnPc
p-radical cation, is +0.8 V versus SCE and, thus, should favor electron transfer to the fullerene triplet excited state. Indeed, excitation of pristine C60 in benzonitrile gives rise to the rapid formation of the fullerene’s diagnostic p-radical anion band. In non-polar solvents, the energy levels for the charge-separated radical pair are, however, significantly raised. As a direct consequence, the electron transfer (reaction 5a) is shut off, while the intriguing energy transfer route (reaction 5b) is still activated. It remains to be shown in the future how the exclusive excitation of the ZnPC chromophore impacts the pathway and formation of the final product.(
3C60) + ZnPc —> (C60•-) + (ZnPc•+) (4a)(
3C60) + ZnPc —> C60 + (3ZnPc) (4b)The oxidative quenching of the fullerene triplet excited state
, leading to the formation of the highly unstable (C60•+) p-radical cation, by electron acceptors (ranging from tetracyanoquinondimethane (TCNQ) and tetracyanoethylene (TCNE) to chloraniline (ClA)) or by cosensitizing the photoinduced electron transfer from a singlet excited N-methylacridinium heaxfluorophosphate (MA) with biphenyl (BP) have been established in solvents of different polarity, e.g., toluene and benzonitrile.2. Functionalized fullerene derivatives
Cycloadditions provide a valuable tool for C
60 functionalization. Usually, 1,2 additions across a [6-6]-ring junction, which possess a higher electron density than the [6-5]-ring junction, are observed. Covalent attachment of a large number of addends across one of the double bonds located at the junctions of two hexagons or across the single bonds at the hexagon-pentagon heterojunction generally affords four different isomeric products. Among these four, the [6-5]-open and the [6-6]-closed are the most interesting isomers. The structure of the [6-5]-open isomer is very appealing, since its fullerene core completely sustains the p-system of pristine C60, but still enables attachment of virtually any functionalizing group. Unfortunately, most of the [6-5]-open derivatives undergo conversion, by thermal, electrochemical, and photochemical initiation, into the thermodynamically more stable [6-6]-closed isomers.In principle, more than just a single addition is possible to the C
60 core since the latter has 30 double bonds, all sharing the same reactivity. Thus, the isolation and characterization of C60 polyadducts renders the C60 framework a useful template for synthesizing novel and structurally complex molecules. For example, when two identical and symmetrical addends are taken up by C60, up to eight isomeric bisadducts can be generated. Their nomenclature refers to the 3-dimensional geometry of the fullerene. In this light, the second addition can take place within either the same hemisphere ("cis"-isomer), at the equator ("equatorial"-isomer) or within the opposite hemisphere ("trans"-isomer).In the following chapters, studies are reviewed that focus on time-resolved and steady-state experiments with functionalized fullerene derivatives. Upfront, a classification should be made to distinguish between the different types of derivatives. The simplest case describes the covalent attachment of addends that lack any VIS absorption and are redox-inactive, in the sense of accepting or donating an electron to the synthetically bound fullerene moiety. It should be emphasized, that these derivatives have evolved as important reference compounds for understanding the intramolecular dynamics within the more complex donor-acceptor dyads. Of increasing complexity are donor-acceptor arrays carrying electroactive addends. For example, in dimethylaniline (DMA), ferrocene (Fc) or tetrathiofulvalene (TTF) based multicomponent supermolecules, such fullerenes are implemented as photosensitizers that are thought to undergo intramolecular electron transfer processes from the adjacent electroactive moiety. Accordingly, these are classified as electroactive but, due to their insignificant VIS absorption characteristics, photoinactive functionalities. Finally, in light of the moderate fullerene absorption features in the VIS region, functionalization of pristine C
60 with chromophoric addends, such as metalloporphyrins or MLCT transition metal complexes, have developed as important objectives. The primary scope of the latter is to promote the VIS absorption characteristics of the resulting dyads and, most importantly, to improve the light harvesting efficiency of the fullerene core. Specifically, suitable photosensitizers assist to extend the fullerene absorption, which, upon excitation, yield energetically high-lying excited states further into the red. As a direct consequence, the role of the fullerene is significantly changed. Under these circumstances, i.e., bearing a photoactive and electroactive moiety, fullerenes operate exclusively as either electron or energy acceptor moieties.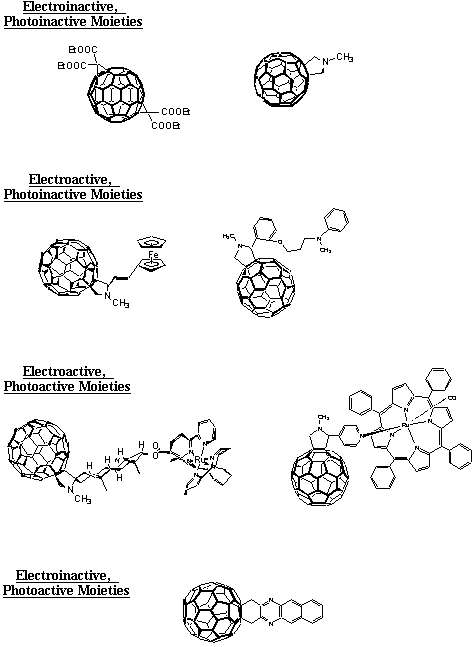
2.1 Electroinactive, photoinactive moieties Converting the fullerene core into a 58
p-electron system greatly alters the electron accepting properties of mono- and multiply functionalized fullerene derivatives. In general, the reduction potentials of most of the known derivatives reveal a shift to more negative potentials. Physico-chemical properties, such as singlet excited states, triplet excited states, reductive quenching by DABCO, transient absorption spectra, and electrochemical reduction, sensitively reflect the functionalization of C60.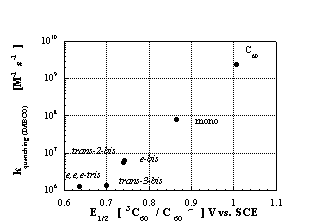
Figure 4: Semilogarithmic plot of the quenching rate constants versus reduction potentials of the triplet excited states of C
60 and various functionalized fullerene derivatives.A representative example is the triplet-triplet absorption, which exhibits a strong blue shift for the e,e,e-(
3C60)[C(COOEt)2]3 of almost 100 nm, relative to pristine C60. This is rationalized to stem from a gradual lifting of the fullerene p-system with increasing number of bis(ethoxycarbonyl)methylene groups. Furthermore, these electronic parameters parallel a slow-down of the triplet excited state quenching with DABCO, amounting to nearly three orders of magnitude for e,e,e-(3C60)[C(COOEt)2]3 as compared to 3C60. A correlation, linking the reductive quenching rates of excited triplet fullerenes with the electrochemically determined redox potentials, shows that functionalization of pristine C60 lowers the ease of reduction in both the ground and in the excited state.A different class of fullerene derivatives, namely, pyrrolidinofullerenes, retain the basic electronic and electrochemical properties of the methanofullerenes along with the features of the added functional groups. These effects impact, for example, the fluorescence and excited state spectra, relative to the methanofullerenes. This change is attributed to the weaker electron donating ability of the adjacent pyrrolidine ring, relative to the cyclopropyl ring.
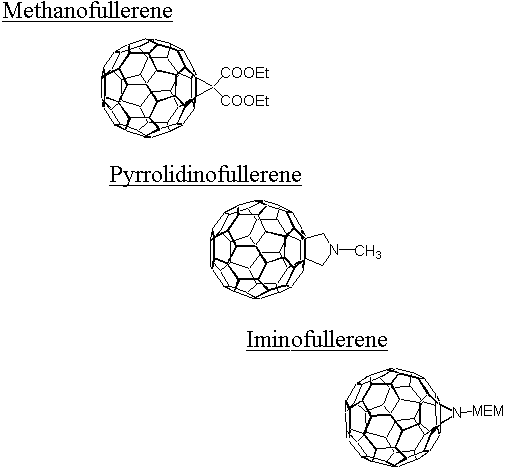
Iminofullerenes have recently attracted attention, since the [6-5]-open structure has gained some degree of stabilization over those of the respective methanofullerene and pyrrolidinofullerene series. Although the [6-6]-closed configuration is still the thermodynamically more stable isomer, the [6-5]-open iminofullerene isomer - in contrast to other [6-5]-open structures - lacks, upon illumination, any evidence of an isomerization reaction. Most importantly, in the [6-5]-open isomer the fullerene core is essentially similar to C
60 and consequently may serve as a promising building block for more complex 3-dimensional systems.A series of spectroscopic properties, concerning the singlet ground state and both excited states, namely, singlet and triplet, further supported by kinetic data, suggests that the electron pair on the nitrogen interacts with the fullerene
p-electrons. This leads, in turn, to a number of important consequences for the electronic structure of the two aziridinofullerene isomers. Considering the free electron pair localized at the nitrogen atom, the [6-6]-closed isomer is best represented by a 60 p-electron system, two of them deriving from the nitrogen and the remaining 58 located within the functionalized fullerene core. In contrast, the [6-5]-open isomer, which, at first glance, may be assumed to be an isoelectronic analogue of pristine C60, carries actually 62 electrons participating in the p-system. In terms of the intriguing electron accepting properties, e.g., ground and excited state, the [6-6]-closed aziridinofullerene reveals, besides a high photostability, not only a better activity than the [6-5]-open isomer, but even outscores the [6-6]-closed methanofullerene and [6-6]-closed pyrrolidinofullerene.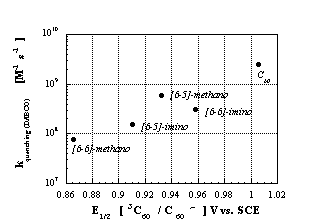
Figure 5: Semilogarithmic plot of the quenching rate constants versus reduction potentials of the triplet excited states of C
60 and various mono-functionalized fullerene isomers.
2.2 Unimolecular Transfer Reactions
2.2.1 Electroactive, photoinactive moieties
Dimethylaniline.
2.2.1 Electroactive, photoinactive moieties Dimethylaniline. The first example of a covalently spaced fullerene
dyad, which was studied under the aspect of electron transfer reactions, employed an
electron donating aniline group via a saturated heterocyclic bridge. In polar
solvents, a significant quenching of the fullerene fluorescence was accompanied by a
strong decrease of the triplet population and, in turn, in the generation of the
charge-separated radical pair. This deactivation route is, however, not observed in
nonpolar solvents, a direct consequence of a less exergonic reaction. Accordingly,
population of the singlet excited state is followed by a conventional quantitative
intersystem crossing to the energetically lower lying triplet excited state. C
C
60-DMA —hn—> (1*C60)-DMA —> (3*C60)-DMA (5b)This promising concept was further extended by increasing the separation between the two redoxactive moieties, e.g., by a 3-
s-bond and a 11-s-bond bridge, with the scope to slow down the back electron transfer. While the earlier configuration, 3-s-bonds, was obtained by replacing the methyl group of the pyrrolidine ring with a N,N,N’,N’-tetramethyl-p-phenylenediamine (TMPD), the latter system implemented the long-ranged separation of DMA via a "norbornylogous" bridge. The strong TMPD donor gives rise to a rapid intramolecular charge separation, irrespective of the solvent polarity. The 11-s-bonds system followed the first class of shorter DMA hybrids. Successful electron transfer occurs, however, with much slower dynamics, and furthermore only in polar solutions. On the other hand, the spatial separation impacts the back electron transfer to yield a considerably life-time of ca. 250 ns. It has been suggested that strong electron coupling enables the fast charge separation, but that the electronic coupling determining the back electron transfer dynamics remains small.
Ferrocene. A different class of electron acceptors, with tunable donor strength, comprised of ferrocenes that can be varied easily via systematic chemical modification. A series of covalently linked fullerene/ferrocene based donor-bridge-acceptor dyads were probed as a function of the nature of the spacer between the donor (ferrocene) and acceptor site (fullerene) and the dielectric constant of the medium, by means of steady-state fluorescence and time-resolved flash photolysis.
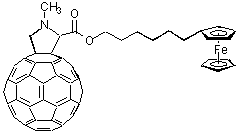
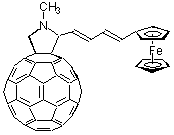
The fullerene fluorescence, a good measure for the singlet excited state life-time, was investigated for several dyads in a variety of solvents of high and low polarity, and was found to be substantially quenched, relative to the plain fullerene model. This suggests rapid intramolecular quenching reactions of the fullerene singlet excited state. Picosecond resolved photolysis of these dyads in toluene showed that the singlet excited state, indeed, undergoes rapid intramolecular quenching, with rate constants ranging between 28 x 10
9 s-1, and 2.3 x 109 s-1 without any evidence for an intersystem crossing to the triplet excited state. Finally, nanosecond-resolved photolysis shows the characteristic fullerene radical-anion band (1055 nm) which confirms the charge separated state with half-lives of up to 2.5 ms in degassed benzonitrile. Remarkably, this applies only for the flexibly spaced ferrocene dyads. In contrast, rigidly spaced dyads fail to disclose any measurable stability of the presumably formed radical pair. The nature of the spacer between C60 and ferrocene, weak electronic ground state interactions, and steady-state fluorescence along with picosecond-resolved photolysis, all suggest two different quenching mechanism: i) through-bond electron transfer for rigid spaced dyads and ii) formation of a transient intramolecular exciplex for flexible dyads.C
60-Fc —hn—> (1*C60)-Fc —> (C60•-)-(Fc•+) (6a)C
60-Fc —hn—> (1*C60)-Fc —> *(C60-Fc) —> (C60•-)-(Fc•+) (6b) (9a)Tetrathiofulvalene. A new family of donor-acceptor fused dyads is based on the linkage of another strong organic electron donor, namely tetrathiofulvalene (TTF), with the fullerene moiety. These compounds have a short and rigid bridge that accounts for some ground state electronic interaction. Systematic introduction of different functional groups at the TTF addend, provide a way for fine tuning the energy gap of the relevant energy levels. The life-time of the triplet excited state relates to the donor strength of the TTF derivative, thus controlling the rate of electron transfer. Interestingly, the life-time of the charge-separated radical pair, resulting from the triplet state quenching, amounts to 79
ms.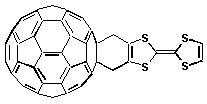
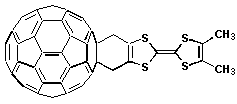
C
60-TTF —hn—> (1*C60)-TTF —> (3*C60)-TTF—> (C60•-)-(TTF•+) (7)
2.2.2 Electroactive, photoactive moieties
In a conceptional extension of the above highlighted results, the linkage of a fullerene to a number of interesting photo- and electroactive species is thought to improve the light harvesting efficiency of the fullerene dyad. More importantly, it changes the function of the fullerene moiety by reducing its role to that of a classical electron acceptor rather than utilizing it also as a photosensitizer. Intramolecular transfer dynamics between the two moieties will be elucidated in this review as a function of the excited state energy of the antenna molecule, donor-acceptor distance, and solvent polarity.
Porphyrin. The initial report of the synthesis and photophysical study of a fullerene linked to a porphyrin pigment was followed by a covalently linked carotenoid-fullerene dyad. The intramolecular dynamics, recorded upon photoexcitation of this carotenoid-fullerene dyad, were studied in detail by, e.g., picosecond and nanosecond resolved photolysis and compared with another porphyrin-fullerene dyad prepared in a different laboratory. The general pattern that stems from these studies is that the porphyrin or carotene singlet excited states decay via singlet energy transfer to the fullerene and/or electron transfer to the fullerene generating a charge-separated state.
P-C
60 —hn—> (1*P)-C60 —> (P•+)-(C60•-) (8a)P-C
60 —hn—> (1*P)-C60 —> P-(1*C60) (8b)Under conditions that the coupling and energetics are favorable, the fullerene first singlet excited state decays by accepting an electron from the adjacent donor to yield the charge-separated state. Using this dyad work as a base, a molecular triad consisting of a diarylporphyrin (P) covalently linked to a carotenoid polyene (C) and a fullerene (C
60) was developed. In 2-methyltetrahydrofuran solution, the photoexcitation of this triad yields the initial C-(P•+)-(C60•-) pair, which subsequently transforms into a (C•+)-P-(C60•-) pair with an overall quantum yield of 0.14. The latter decays via charge recombination to afford the carotenoid triplet state, rather than the ground state, with a time constant of 170 ns. Time-resolved EPR spectroscopy disclosed detection of the spin-polarized radical pair and carotenoid triplet states, substantiating that the carotenoid triplet indeed evolves from the (C•+)-P-(C60•-) pair by radical recombination. The generation of a long-lived charge separated state in such a triad system as product of photoinduced electron transfer and formation of a triplet state via charge recombination are phenomena heretofore observed almost exclusively in photosynthetic reaction centers.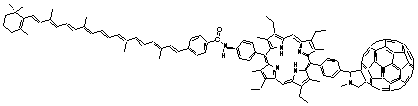
C-P-C
60 —hn—> C-(1*P)-C60 —ET—> C-(P•+)-(C60•-)—ET—
> (C•+)-P-(C60•-) —BET—> (3*C)-P-C60 (9)A different development, specifically regarding the linkage of a porphyrin antenna molecule with the electron accepting fullerene moiety, encompasses the convergent synthesis of several different fullerene hybrids with tunable molecular topologies. In these intriguing hybrids polyether chains are used as a linking block to provide effects which are based on the complexation of metal cations, such as potassium. This is thought to bring the two moieties closer together and promote the intramolecular electron transfer dynamics. Electrochemical, photophysical, computational, and NMR studies, indeed, provide remarkable evidence for a complexation of potassium cations which alters the fullerene porphyrin distance and substantially accelerates the intramolecular dynamics.
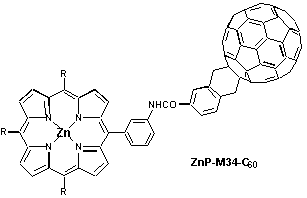
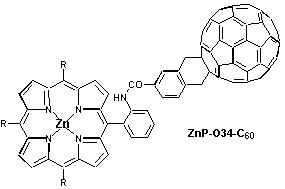
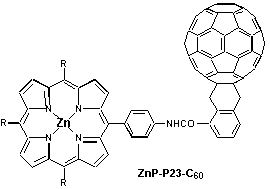
Introduction of a phenyl spacer between the electron accepting fullerene and the photosensitizer / electron donor zinc porphyrin, enabled the systematic variation of the linkage, e.g., by positioning it in the ortho-, meta- or para-position of the phenyl ring. Regardless of the linkage position, the rapidly occurring charge separation, following the initial photoexcitation, was always succeeded by a charge recombination which results in the complete restoration of the ground state. While in polar THF the radical pair evolves from the singlet excited state of both moieties, fullerene and porphyrin, the situation is different in benzene where the singlet excited states efficiently mix and, as a consequence, energy transfer dominates over electron transfer. In light of stabilizing the charge-separated radical pair, it is important to note that both charge separation and charge recombination, are particularly slowed-down by meta-linkage.
The successful design of another sequential electron transfer relay system has been achieved when a porphyrin fullerene donor acceptor pair is spaced with a pyromellitimide functionality. Starting with the initial excited porphyrin state the imide moiety is found to accept an electron from the ZnTPP chromophore affording a charge-separated radical pair. This transient state subsequently transfers the accepted charge from the imide to the end terminus, namely, the fullerene moiety, forming a spatially separated (P•+)-In-(C
60•-) radical pair. The resulting charge separated state undergoes a remarkably slow back electron transfer with a rate of 7.7 x 108 s-1 and an overall quantum yield of only 0.46.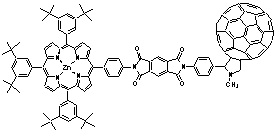
P-In-C
60 —hn—> (1*P)-In-C60 —ET—> (P•+)-(In•-)-C60—ET—
> (P•+)-In-(C60•-) —BET—> P-In-C60 (10)Enhancement of the interactions between the fullerene and porphyrin units has alternatively been achieved by linking two fullerenes units, rather than one, to the meso-position of a macrocyclic porphyrin ligand. The results from emission experiments fall well in line with the expectation that introduction of a second electron acceptor promotes the efficiency of chromophore deactivation and, in turn, electron transfer relative to a conventional dyad.
Ruthenium(II) complex. The strong emission and long life-time of the ruthenium metal-to-ligand charge transfer states, generated with high quantum yields, render them attractive starting points for energy and electron transfer processes. The energetically high-lying ruthenium excited MLCT state (E = 1.96 eV) should, in principle, facilitate photoinduced intramolecular electron transfer to suitable acceptors, such as the discussed fullerenes. This inspired researchers to employ ruthenium(II) complexes in the design of light-harvesting devices and to investigate them in electron transfer reactions.
The first study, following this concept, reported evidence for photoinduced electron transfer from the MLCT excited state of a covalently, but flexibly, attached ruthenium(II) complex, namely, [Ru(byp)
3]2+ to the fullerene moiety. In a thin solid film, a photoinduced electron spin resonance signal was noted, which exhibits signatures of both the oxidized ruthenium(III) complex and the reduced C60 p-radical anion. The life-time for this state is reported to be on the order of milliseconds at 80 K.[Ru(byp)
3]2+-C60 —hn—> *[Ru(byp)3]2+-C60—ET—> [Ru(byp)3]3+-(C60•-) (11)
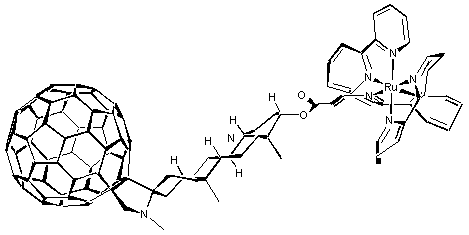
In an extension of the former work, the principles of the 1,3-dipolar cycloaddition reaction of azomethine ylides to C60 has recently been applied to prepare a pyrrolidinofullerene, rigidly linked via an androstane spacer to a ruthenium(II) trisbipyridine chromophore. Steady-state fluorescence and time-resolved flash photolytic investigations of this ruthenium(II)-androstane-C60 dyad focused on the variation of the dielectric constant of the medium. In CH2Cl2, and CH3CN the emission was substantially quenched, relative to a ruthenium(II) trisbipyridine model, suggesting intramolecular quenching of the ruthenium MLCT excited state. Picosecond-resolved photolysis of the dyad shows that the light-induced formation of the photoexcited ruthenium center is followed by rapid intramolecular quenching. Nanosecond-resolved photolysis confirmed the formation of the characteristic fullerene p-radical anion band at lmax = 1040 nm and life-times of the charge separated state of 304 ns and 145 ns in CH2Cl2 and CH3CN, respectively, were observed.
Rotaxane. This novel self-assembling three component array consists of a macrocycle coordinating ring, a redoxactive copper(I) center and a bisfunctionalized fragment. The resulting complex, with the bisfunctionalized fragment threaded inside the ring, was reacted with a monofunctionalized fullerene derivative to afford a soluble rotaxane with two fullerenes as end-terminating stoppers. Both excited states, the MLCT state of the macrocyclic copper(I) complex and that of the fullerene moiety, are substantially quenched. The outcome of this rapid intramolecular deactivation is, in view of the excited state energies, not surprisingly very different in character and product formation. For example, deactivation of the fullerene excited state follows an energy transfer to the adjacent Cu(I) complex. The excited state of the latter is, in contrast, mainly quenched by electron transfer to form the charge-separated radical pair comprised of Cu(II) and the one-electron reduced fullerene
p-radical anion (C60•-).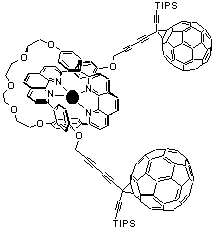
2.1.3 Electroinactive, photoactive moieties
Arenes and Pyrazines. Fluorophore-fullerene adducts, such as, anthracene and pyrene derivatives, have been prepared via Diels-Alder addition reactions employing electron rich silylbutadienes and fullerenes as precursor. An energy transfer route has been proposed as the major deactivation pathway of these bichromophoric systems, generating the long-lived and highly reactive fullerene triplet excited state.
Arene-C
60 —hn—> (1*Arene)-C60—> Arene-(1*C60) (12)
In an alternative approach to design arene-fullerene systems, a thermal reaction of C60 with dihydrocyclobutaarenes was pursued, affording dyads that contain a series of arenes ranging from benzene to pyrene. The quantitative fluorescence quenching of the attached aromatic arenes has been ascribed to a very rapid intramolecular singlet-singlet energy transfer that transforms the short-lived and moderately redox-active excited states of the investigated arenes into the highly reactive fullerene triplet excited state.
Functionalization of pristine C60 with several different pyrazine derivatives, carrying substituents with different electron-inducing abilities is another promising strategy to improve the UV-VIS absorption characteristics and, in turn, the light harvesting efficiency of the resulting dyads, relative to pristine C60. Photoexcitation of the pyrazine moieties in these dyads leads to the formation of their singlet excited states. In contrast to the pyrazine models, but in analogy to the fullerene arene dyads, photoexcitation of all the fullerene pyrazine dyads is followed by rapid intramolecular deactivation processes via energy transfer to the fullerene ground state with half-lives ranging typically between 37 and 100 ps.
Acknowledgment
This work was supported by the Office of Basic Energy Sciences of the U.S. Dept. of Energy. This is contribution No. NDRL-4101 from the Notre Dame Radiation Laboratory
.Table 1: Ground State and Excited State Properties of C
60, C70, and a Monofunctionalized Fullerene Derivative, C60C(COOEt)2.Property |
C 60a |
C 70b |
C 60C(COOEt)2c |
E (Singlet) |
1.99 eV |
1.90 eV |
1.79 eV |
E (Triplet) |
1.57 eV |
1.60 eV |
1.502 eV |
l max |
920 nm |
660 nm |
920 nm |
l max |
747nm |
400 nm |
720 nm |
e (Triplet) |
20 000 M-1cm-1 |
14 000 M-1cm-1 |
14 000 M-1cm-1 |
t (Singlet) |
1.3 ns |
0.7 ns |
2.23 ns |
t (Triplet) |
135 ms |
11.8 ms |
40 ms |
E 1/2 1C60/(C60•-) |
1.44 V vs SCE |
1.39 vs SCE |
1.16 vs SCE |
E 1/2 3C60/(C60•-) |
1.01 vs SCE |
1.09 vs SCE |
0.86 vs SCE |
F (Fluorescence) |
1.0 x 10 -4 |
3.7 x 10 -4 |
6.0 x 10 -4 |
F (Triplet) |
0.96 |
0.9 |
0.96 |
k q (oxygen) |
1.6 x 10 9 M-1s-1 |
1.9 x 10 9 M-1s-1 |
1.8 x 10 9 M-1s-1 |
k q (DABCO) |
2.5 x 10 9 M-1s-1 |
7.7 x 10 7 M-1s-1 |
|
k ET (biphenyl) |
1.7 x 10 10 M-1s-1 |
2.0 x 10 10 M-1s-1 |
1.8 x 10 10 M-1s-1 |
a
26,40; b 26,135; c 105,106Table 2: Rate Constant for the Reductive Quenching of C
60 and C70Quenching Donor |
C 60toluene; k q x 109 M-1s-1 |
C 60benzonitrile; k q x 109 M-1s-1 |
C 70benzonitrile; k q x 109 M-1s-1 |
Tetramethyphenyldiamine |
5.2 m |
5.1 o |
|
N,N-Diethylaniline |
5.1 m |
7.1 o |
|
N.N-Dimethylaniline |
2.6 m |
4.8 o |
|
Diphenylamine |
0.87 m |
3.5 o |
|
Triphenylamine |
0.89 m |
1.4 o |
|
Tripropylamine |
0.94 m |
||
Triethylamine |
2.9 d |
0.96 m |
0.95 o |
Aniline |
0.22 m |
0.15 o |
|
Dibutylamine |
0.0072 m |
0.1 o |
|
Dipropylamine |
0.0065 m |
0.089 o |
|
Diethylamine |
0.11 o |
||
Dibenzylamine |
0.019 o |
||
Pyrene |
0.00012 o |
||
Diazabicyclooctane |
2.5 e |
0.18 e |
|
Polysilane |
0.21 n |
||
Ferrocene |
3.3 f |
||
Tetrathiafulvalene |
9.9 g |
5.0 g |
|
BEDT-Tetrathiafulvalene |
5.9 g |
3.8 g |
|
b -Carotene |
7.8 h |
5.3 h |
|
Tetramethylbenzidine |
6.7 i |
1.3 i |
|
Zn Phthalocyanine |
2.1 j |
1.6 j |
|
H 2 Phthalocyanine |
2.8 j |
3.0 j |
|
Quaterthiophene |
0.31 k |
||
TEMPO |
3.3 c; l |
||
HTEMPO |
1.9 c; l |
||
DTBN |
3.0 c |
a
(n-propanol); b (CH3CN, BZCN); c (CH2Cl2); d ; e ; f ; g ; h ; i ; j ; k ; l m ; n ; oTable 3:
Table 3: Photophysical Properties of cis-3-C
60[C(COOEt)2]2, e-C60[C(COOEt)2]2, trans-3-C60[C(COOEt)2]2, trans-2-C60[C(COOEt)2]2, and e,e,e-C60[C(COOEt)2]3.
Property |
cis-3- C 60[C(COOEt)2]2
|
e- C 60[C(COOEt)2]2
|
trans-3- C 60[C(COOEt)2]2
|
trans-2- C 60[C(COOEt)2]2
|
e,e,e- C 60[C(COOEt)2]3
|
E (Singlet) |
1.775 eV |
1.765 eV |
1.770 eV |
1.765 eV |
|
E (Triplet) |
1.50 eV |
1.502 eV |
1.504 eV |
1.498 eV |
|
l max |
885 nm |
895 nm |
875 nm |
868 nm |
|
l max |
690 nm |
710 nm |
705 nm |
690 nm |
650 nm |
e (Triplet) |
8 400 M-1cm-1 |
9 700 M-1cm-1 |
8 000 M-1cm-1 |
8 200 M-1cm-1 |
7 500 M-1cm-1 |
t (Singlet) |
4.2 ns |
2.8 ns |
2.4 ns |
4.52 ns |
|
E 1/2 (Singlet) |
1.02 V vs SCE |
0.97 V vs SCE |
1.01 vs SCE |
0.91 vs SCE |
|
E 1/2 (Triplet) |
0.74 vs SCE |
0.70 V vs SCE |
0.74 vs SCE |
0.64 vs SCE |
|
k q (DABCO) |
6.4 x 10 6 M-1s-1 |
1.4 x 10 6 M-1s-1 |
5.8 x 10 6 M-1s-1 |
1.3 x 10 6 M-1s-1 |
References
1)Kroto, H. W.; Heath, J. R.; O'Brien, S. C.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162-164.
2)Krätschmer, W.; Lamb, L. D.; Fostiropoulos, K.; Huffman, D. R. Nature 1990, 347, 354-358.
3)Haddon, R. C.; Siegrist, T.; Fleming, R. M.; Bridenbaugh, P. M.; Laudise, R. A. J. Mater. Chem. 1995, 5, 1719-1724.
4)Haddon, R. C.; Perel, A. S.; Morris, R. C.; Plastra, T. T. M.; Hebrad, A. F.; Fleming, R. M. Appl. Phys. Lett. 1995, 67, 121-123.
5)Haddon, R. C.; Hebard, A. F.; Rosseinsky, M. J.; Murphy, D. W.; Duclos, S. J.; Lyons, K. B.; Miller, B.; Rosamilia, J. M.; Fleming, R. M.; Kortan, A. R.; Glarum, S. H.; Makhija, A. V.; Muller, A. J.; Eick, R. H.; Zahurak, S. M.; Tycko, R.; Dabbagh, G.; Thiel, F. A. Nature 1991, 350, 320-322.
6)Haddon, R. C. Acc. Chem. Res. 1988, 21, 243-249.
7)Mauser, H.; Hommes, N. J. R. v. E.; Clark, T.; Hirsch, A.; Pietzak, B.; Weidinger, A.; Dunsch, L. Angew. Chem. Int. Ed. Engl. 1997, 36, 2835-2838.
8)Haddon, R. C.; Brus, L. E.; Raghavachari, K. Chem. Phys. Lett. 1986, 125, 459.
9)Dubois, D.; Kadish, K. M.; Flanagan, S.; Wilson, L. J. J. Am. Chem. Soc. 1991, 113, 7773-7774.
10)Dubois, D.; Kadish, K. M.; Flanagan, S.; Haufler, R. E.; Chibante, L. P. F.; Wilson, L. J. J. Am. Chem. Soc. 1991, 113, 4364-4366.
11)Allemand, P.-M.; Koch, A.; Wudl, F.; Rubin, Y.; Diederich, F.; Alvarez, M. M.; Anz, S. J.; Whetten, R. L. J. Am. Chem. Soc. 1991, 113, 1051-1052.
12)Xie, Q.; Perez-Cordero, E.; Echegoyen, L. J. Am. Chem. Soc. 1992, 114, 3978-80.
13)Guldi, D. M.; Asmus, K.-D. J. Am. Chem. Soc. 1997, 119, 5744-5.
14)Imahori, H.; Hagiwara, K.; Akiyama, T.; Aoki, M.; Taniguchi, S.; Okada, T.; Shirakawa, M.; Sakata, Y. Chem. Phys. Lett. 1996, 263, 545-550.
15)Kato, T.; Kodama, T.; Shida, T.; Nakagawa, T.; Matsui, Y.; Suzuki, S.; Shiromaru, H.; Yamauchi, K.; Achiba, Y. Chem. Phys. Lett. 1991, 180, 446-50.
16)Yang, Y.; Arias, F.; Echegoyen, L.; Chibante, L. P.; Flanagan, S.; Robertson, A.; Wilson, L. J. J. Am. Chem. Soc. 1995, 117, 7801-7804.
17)Hirsch, A. Synthesis 1995, 895.
18)Prato, M.; Maggini, M. Acc. Chem. Res. 1998, 31, 00000.
19)Prato, M. J. Mater. Chem. 1997, 7, 1097-1109.
20)Diederich, F.; Thilgen, C. Science 1996, 271, 317-323.
21)Diederich, F. Pure & Apl. Chem. 1997, 69, 395-400.
22)Taylor, R.; Walton, D. R. M. Nature 1993, 363, 685-693.
23)Sun, Y.-P., Ed.; Marcel Dekker, New York, 1997; Vol. 1.
24)Prato, M. J. Mater. Chem.1997, 7, 1097-109.
25)Imahori, H.; Yamada, K.; Hasegawa, M.; Taniguchi, S.; Okada, T.; Sakata, Y. Angew. Chem. Int. Ed. Engl. 1997, 36, 2626-9.
26)Foote, C. S. Top. Curr. Chem. 1994, 169, 347-363.
27)Leach, S.; Vervloet, M.; Despres, A.; Brcheret, E.; Hare, P.; Dennis, T. J.; Kroto, W.; Taylor, R.; Walton, D. R. M. Chem. Phys. 1992, 160, 451-466.
28)Ajie, H.; Alvarez, M. M.; Anz, S. J.; Beck, R. D.; Diederich, F.; Fostiropoulos, K.; Huffman, D. R.; Kraetschmer, W.; Rubin, Y.; Schriver, K. E.; Sensharma, D.; Whetten, R. L. J. Phys. Chem. 1990, 94, 8630-3.
29)Sun, Y.-P.; Wang, P.; Hamilton, N. B. J. Am. Chem. Soc. 1993, 115, 6378-6381.
30)Wang, Y. J. Phys. Chem. 1992, 96, 764-767.
31)Sibley, S. P.; Argentine, S. M.; Francis, A. H. Chem. Phys. Lett. 1992, 188, 187-93.
32)Zeng, Y.; Biczok, L.; Linschitz, H. J. Phys. Chem. 1992, 96, 5237-5239.
33)Ebbesen, T. W.; Tanigaki, K.; Kuroshima, S. Chem. Phys. Lett. 1991, 181, 501-4.
34)Ebbesen, T. W.; Tanigaki, K.; Kuroshima, S.; Hiura, H.; Takahashi, H. Springer Proc. Phys. 1992, 284-8.
35)Palit, D. K.; Sapre, A. V.; Mittal, J. P.; Rao, C. N. R. Chem. Phys. Lett. 1992, 195, 1-6.
36)Wasielewski, M. R.; O'Neil, M. P.; Lykke, K. R.; Pellin, M. J.; Gruen, D. M. J. Am. Chem. Soc. 1991, 113, 2774-6.
37)Sension, R. J.; Phillips, C. M.; Szarka, A. Z.; Romanow, W. J.; McGhie, A. R.; McCauley, J. P. J.; Smith, A. B. I.; Hochstrasser, R. M. J. Phys. Chem. 1991, 95, 6075-8.
38)Lee, M.; Song, O. K.; Seo, J. C.; Kim, D.; Suh, Y. D.; Jin, S. M.; Kim, S. K. Chem. Phys. Lett. 1992, 196, 325-9.
39)Tanigaki, K.; Ebbesen, T. W.; Kuroshima, S. Chem. Phys. Lett. 1991, 185, 189-92.
40)Arbogast, J. W.; Darmanyan, A. P.; Foote, C. S.; Rubin, Y.; Diederich, F. N.; Alvarez, M. M.; Anz, S. J.; Whetten, R. L. J. Phys. Chem. 1991, 95, 11-12.
41)Sension, R. J.; Szarka, A. Z.; Smith, G. R.; Hochstrasser, R. M. Chem. Phys. Lett. 1991, 185, 179-83.
42)Kajii, Y.; Nakagawa, T.; Suzuki, S.; Achiba, Y.; Obi, K.; Shibuya, K. Chem. Phys. Lett. 1991, 181, 100-104.
43)Biczok, L.; Linschitz, H.; Walter, R. I. Chem. Phys. Lett. 1992, 195, 339-46.
44)Dimitrijevic, N. M.; Kamat, P. V. J. Phys. Chem. 1992, 96, 4811-4814.
45)Palit, D. K.; Sapre, A. V.; Mittal, J. P. Indian J. Chem., Sect. A 1992, F46.
46)Bensasson, R. V.; Hill, T. J.; Lambert, C.; Land, E. J.; Leach, S.; Truscott, T. G. Chem. Phys. Lett. 1993, 206, 197-202.
47)Ausman, K. D.; Weisman, R. B. Res. Chem. Intermed. 1997, 23, 431-451.
48)Etheridge, H. T., III; Averitt, R. D.; Halas, N. J.; Weisman, R. B. J. Phys. Chem. 1995, 99, 11306-11308.
49)Fraelich, M. R.; Weisman, R. B. J. Phys. Chem. 1993, 97, 11145-11147.
50)Guldi, D. M.; Hungerbühler, H.; Asmus, K.-D. J. Phys. Chem. A 1997, 101, 1472-1481.
51)Ito, O. Res. Chem. Intermed. 1997, 23, 389-402.
52)Arbogast, J. W.; Foote, C. S.; Kao, M. J. Am. Chem. Soc. 1992, 114, 2277-2279.
53)Guldi, D. M.; Huie, R. E.; Neta, P.; Hungerbühler, H.; Asmus, K.-D. Chem. Phys. Lett. 1994, 223, 511-516.
54)Guldi, D. M.; Maggini, M.; Scorrano, G.; Prato, M. Res. Chem. Intermed. 1997, 23, 561-573.
55)Guldi, D. M.; Maggini, M.; Scorrano, G.; Prato, M. J. Am. Chem. Soc. 1997, 119, 974-980.
56)Alam, M. M.; Watanabe, A.; Ito, O. J. Photochem. Photobiol. A: Chem. 1997, 104, 59-64.
57)Sasaki, Y.; Yoshikawa, Y.; Watanabe, A.; Ito, O. J. Chem. Soc., Faraday Trans 1995, 91, 2287-2290.
58)Ito, O.; Sasaki, Y.; Yoshikawa, Y.; Watanabe, A. J. Phys. Chem. 1995, 99, 9838-9842.
59)Samanta, A.; Kamat, P. V. Chem. Phys. Lett. 1992, 199, 635-639.
60)Schaffner, E.; Fischer, H. J. Phys. Chem 1993, 97, 13149-13151.
61)Watanabe, A.; Ito, O. J. Phys. Chem. 1994, 98, 7736-7740.
62)Bennati, M.; Grupp, A.; Bäuerle, P.; Mehring, M. Chem. Phys. 1994, 185, 221-227.
63)Bennati, M.; Grupp, A.; Bäuerle, P.; Mehring, M. Mol. Cryst. Liq. Cryst. 1994, 256, 751-756.
64)Sun, Y.-P.; Ma, B.; Lawson, G. E. Chem. Phys. Lett. 1995, 233, 57-62.
65)Ma, B.; Lawson, G. E.; Bunker, C. E.; Kitaygorodskiy, A.; Sun, Y.-P. Chem. Phys. Lett. 1995, 247, 51-56.
66)Wang, Y. J. Phys. Chem. 1992, 96, 764-767.
67)Ghosh, H. N.; Pal, H.; Sapre, A. V.; Mittal, J. P. J. Am. Chem. Soc. 1993, 115, 11722-11727.
68)Scurlock, R. D.; Ogilby, P. R. J. Photochem. Photobiol. A: Chem. 1995, 91, 21-25.
69)Nojiri, T.; Alam, M. M.; Konami, H.; Watanabe, A.; Ito, O. J. Phys. Chem. A 1997, 101, 7943-7947.
70)Sasaki, Y.; Fujitsuka, M.; Watanabe, A.; Ito, O. J. Chem. Soc., Faraday Trans. 1997, 93, 4275-4279.
71)Nadtochenko, V. A.; Denisov, N. N.; Rubtsov, I. V.; Lobach, A. S.; Moravsky, A. P. Russ. Chem. Bull. 1993, 42, 1171-1173.
72)Nadtochenko, V. A.; Denisov, N. N.; Lobach, A. S.; Moravskii, A. P. Zh. Fiz. Khim. 1994, 68, 228-231.
73)Nadtochenko, V. A.; Denisov, N. N.; Rubtsov, I. V.; Lobach, A. S.; Moravskii, A. P. Chem. Phys. Lett. 1993, 208, 431-435.
74)Michaeli, S.; Meiklyar, V.; Schulz, M.; Möbius, K.; Levanon, H. J. Phys. Chem. 1994, 98, 7444-7447.
75)Michaeli, S.; Meiklyar, V.; Endeward, B.; Möbius, K.; Levanon, H. Res. Chem. Intermed. 1997, 23, 505-517.
76)Steren, C. A.; Van Willigen, H. Proc. - Indian Acad. Sci., Chem. Sci. 1994, 106, 1671-1679.
77)Steren, C. A.; Levstein, P. R.; van Willigen, H.; Linschitz, H.; Biczok, L. Chem. Phys. Lett. 1993, 204, 23-8.
78)Steren, C. A.; van Willigen, H.; Biczók, L.; Gupta, N.; Linschitz, H. J. Phys. Chem. 1996, 100, 8920-8926.
79)Nonell, S.; Arbogast, J. W.; Foote, C. S. J. Phys. Chem. 1992, 96, 4169-4170.
80)Safonov, I. G.; Courtney, S. H.; Schuster, D. I. Res. Chem. Intermed. 1997, 23, 541-548.
81)Thilgen, C.; Herrmann, A.; Diederich, F. Angew. Chem. Int. Ed. Engl. 1997, 36, 2268-2280.
82)Hirsch, A. The Chemsitry of the Fullerenes; Georg Thieme Verlag: Stuttgart, 1994.
83)Hirsch, A. Synthesis 1995, 895-913.
84)Walbiner, M.; Fischer, H. J. Phys. Chem. 1993, 97, 4880-4881.
85)Williams, R. M.; Koeberg, M.; Lawson, J. M.; An, Y.-Z.; Rubin, Y.; Paddon-Row, M. N.; Verhoeven, J. W. J. Org. Chem. 1996, 61, 5055-5062.
86)Williams, R. M.; Zwier, J. M.; Verhoeven, J. W. J. Am. Chem. Soc. 1995, 117, 4093-4099.
87)Llacay, J.; Veciana, J.; Vidal-Gancedo, J.; Bourdelande, J. L.; Gonzalez-Moreno, R.; Rovira, C. J. Org. Chem. 1998, 63, 5201-5210.
88)Dietel, E.; Hirsch, A.; Zhou, J.; Rieker, A. J. Chem. Soc., Perkin Trans. 2 1998, 1357-1364.
89)Baran, P. S.; Monaco, R. R.; Khan, A. U.; Schuster, D. I.; Wilson, S. R. J. Am. Chem. Soc. 1997, 119, 8363-8364.
90)Higashida, S.; Imahori, H.; Kaneda, T.; Sakata, Y. Chem. Lett. 1998, 605-606.
91)Imahori, H.; Hagiwara, K.; Akiyama, T.; Taniguchi, S.; Okada, T.; Sakata, Y. Chem. Lett. 1995, 265-266.
92)Akiyama, T.; Imahori, H.; Ajawakom, A.; Sakata, Y. Chem. Lett. 1996, 907-908.
93)Bell, T. D. M.; Smith, T. A.; Ghiggino, K. P.; Ranasinghe, M. G.; Shephard, M. J.; Padden-Row, M. N. Chem. Phys. Lett. 1997, 268, 223-228.
94)Imahori, H.; Hagiwara, K.; Aoki, M.; Akiyama, T.; Taniguchi, S.; Okada, T.; Shirakawa, M.; Sakata, Y. J. Am. Chem. Soc. 1996, 118, 11771-11782.
95)Kuciauskas, D.; Lin, S.; Seely, G. R.; Moore, A. L.; Moore, T. A.; Gust, D.; Drovetskaya, T.; Reed, C. A.; Boyd, P. D. W. J. Phys. Chem. 1996, 100, 15926-15932.
96)Liddell, P. A.; Kuciauskas, D.; Sumida, J. P.; Nash, B.; Nguyen, D.; Moore, A. L.; Moore, T. A.; Gust, D. J. Am. Chem. Soc. 1977, 119, 1400-1405.
97)Liddell, P. A.; Sumida, J. P.; Macpherson, A. N.; Noss, L.; Seely, G. R.; Clark, K. N.; Moore, A. L.; Moore, T. A.; Gust, D. Photochem. Photobiol. 1994, 60, 537-541.
98)Sun, Y.; Drovetskaya, T.; Bolskar, R. D.; Bau, R.; Boyd, P. D. W.; Reed, C. A. J. Org. Chem. 1997, 62, 3642-3649.
99)Shephard, M. J.; Paddon-Row, M. N. Aust. J. Chem. 1996, 49, 395-403.
100)Bell, T. D. M.; Smith, T. A.; Ghiggino, K. P.; Ranasinghe, M. G.; Shephard, M. J.; Paddon-Row, M. N. Chem. Phys. Lett. 1997, 268, 223-228.
101)Maggini, M.; Dono, A.; Scorrano, G.; Prato, M. J. Chem. Soc., Chem. Commun. 1995, 845-846.
102)Armspach, D.; Constable, E. C.; Diederich, F.; Housecraft, C. E.; Nierengarten, J.-F. J. Chem. Soc., Chem. Commun. 1996, 2009-2010.
103)Issacs, L.; Diederich, F. Helv. Chim. Acta 1993, 76, 2454-2464.
104)Brezova, V.; Stasko, A.; Rapta, P.; Guldi, D. M.; Asmus, K.-D.; Dinse, K.-P. Magn. Reson. Chem. 1997, 35, 795-801.
105)Guldi, D. M.; Hungerbühler, H.; Asmus, K. D. J. Phys. Chem. 1995, 99, 9380-9385.
106)Guldi, D. M.; Asmus, K.-D. J. Phys. Chem. A 1997, 101, 1472-1481.
107)Zhou, D.; Tan, H.; Gan, L.; Luo, C.; Huang, C.; Yao, G.; Zhang, P. Chem. Lett. 1995, 649-650.
108)Zhou, D.; Gan, L.; Tan, H.; Luo, C.; Huang, C.; Yao, G.; Zhang, B. J. Photochem. Phogobiol. A: Chem. 1996, 99, 37-43.
109)Lin, S.-K.; Shiu, L.-L.; Chien, K.-M.; Luh, T.-Y.; Lin, T.-I. J. Phys. Chem. 1995, 99, 105-111.
110)Ma, B.; Bunker, C. E.; Guduru, R.; Zhang, X.-F.; Sun, Y.-P. J. Phys. Chem. A 1997, 101, 5626-32.
111)Bensasson, R. V.; Bienvenue, E.; Fabre, C.; Janot, J.-M.; Land, E. J.; Leach, S.; Leboulaire, V.; Rassat, A.; Roux, S.; Seta, P. Chem. Eur. J. 1998, 4, 270-278.
112)Guldi, D. M.; Maggini, M. Gazz. Chim. Ital. 1997, 127, 779-785.
113)Luo, C.; Fujitsuka, M.; Watanabe, A.; Ito, O.; Gan, L.; Huang, Y.; Huang, C.-H. J. Chem. Soc., Faraday Trans. 1998, 94, 527-32.
114)George Thomas, K.; Biju, V.; George, M. V.; Guldi, D. M.; Kamat, P. V. J. Phys. Chem. A 1998, 102, 5341-5348.
115)Anderson, J. L.; An, Y.-Z.; Rubin, Y.; Foote, C. S. J. Am. Chem. Soc. 1994, 116, 9763-9764.
116)Prato, M.; Li, Q.; Wudl, F.; Luchini, V. J. Am. Chem. Soc. 1993, 115, 1148-1150.
117)Prato, M.; Li, Q. C.; Wudl, F.; Lucchini, V. J. Am. Chem. Soc. 1993, 115, 1148-1150.
118)Guldi, D. M.; Maggini, M. J. Phys. Chem. 1998, submitted.
119)Williams, R. M.; Zwier, J. M.; Verhoeven, J. W. J. Am. Chem. Soc. 1995, 117, 4093-4099.
120)George Thomas, K.; Biju, V.; Guldi, D. M.; George, M. V.; Kamat, P. V. Chem. Commun. 1998, submitted.
121)Diekers, M.; Hirsch, A.; Pyo, S.; Rivera, J.; Echegoyen, L. Eur. J. Org. Chem. 1998, 1111-1121.
122)Gust, D.; Moore, T. A.; Moore, A. L. Res. Chem. Intermed. 1997, 120, 4398-4405.
123)Liddell, P. A.; Kuciauskas, D.; Sumida, J. P.; Nash, B.; Nguyen, D.; Moore, A. L.; Moore, T. A.; Gust, D. J. Am. Chem. Soc. 1997, 119, 1400-1405.
124)Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; Zelewsky, A. Coord. Chem. Rev. 1988, 84, 85.
125)Balzani, V.; Juris, A.; Venturi, M.; Campagna, S.; Serroni, S. Chem. Rev. 1996, 96, 759.
126)Sariciftci, N. S.; Wudl, F.; Heeger, A. J.; Maggini, M.; Scorrano, G.; Prato, M.; Bourassa, J.; Ford, P. C. Chem. Phys. Lett. 1995, 247, 510-514.
127)Maggini, M.; Guldi, D. M.; Mondini, S.; Scorrano, G.; Paolucci, F.; Ceroni, P.; Roffia, S. Chem. Eur. J. 1998, 4, 1992-2000.
128)Maggini, M.; Guldi, D. M.; Mondini, S.; Scorrano, G.; Paolucci, F.; Ceroni, P.; Roffia, S. J. Phys. Chem. A 1998, 102, submitted.
129)Armspach, D.; Constable, E. C.; Diederich, F.; Housecroft, C. E.; Nierengarten, J.-F. Chem. Eur. J. 1998, 4, 723-733.
130)Armaroli, N.; Diederich, F.; Dietrich-Buchecker, C. O.; Flamigni, L.; Marconi, G.; Nierengarten, J.-F.; Sauvage, J.-P. Chem. Eur. J. 1998, 4, 406-416.
131)Gareis, T.; Köthe, O.; Daug, J. Eur. J. Org. Chem. 1998, 1549-1557.
132)Nakamura, Y.; Minowa, T.; Tobita, S.; Shizuka, H.; Nishimura, J. J. Chem. Soc., Perkin Trans. 2 1995, 2351-2357.
133)Nakamura, Y.; Minowa, T.; Hayashida, Y.; Tobita, S.; Shizuka, H.; Nishimura, J. J. Chem. Soc, Faraday Trans. 1996, 92, 377-382.
134)Guldi, D. M.; Torres-Garcia, G.; Mattay, J. J. Phys. Chem. A 1998, 102, in press.
135)Arbogast, J. S.; Foote, C. S. J. Am. Chem. Soc. 1991, 113, 8886-8889.
136)Sun, Y.-P.; Ma, B.; Lawson, G. E. Chem. Phys. Lett. 1995, 233, 57-62.
137)Sasaki, Y.; Yoshikawa, Y.; Watanabe, A.; Ito, O. J. Chem. Soc., Faraday Trans. 1995, 91, 2287-2290.
138)Osaki, T.; Tai, Y.; Tazawa, M.; Tanemura, S.; Inukai, K.; Ishiguro, K.; Sawaki, Y.; Saito, Y.; Shinohara, H.; Nagashima, H. Chem. Lett. 1993, 789-792.