| Papers and Posters | Site Home Page |
Organic azides photolysis in the crystalline state: the mechanism of polymer formation.
S.V. Zelentsov, A.V.Oleinik
The Chemical Department, Nizhnii Novgorod University, Gagarin Ave., 23.
Nizhnii Novgorod 603600 Russia
E-mail: zelen@ichem.unn.runnet.ru
Summary.
The Chemical Department, Nizhnii Novgorod University, Gagarin Ave., 23.
Nizhnii Novgorod 603600 Russia
E-mail: zelen@ichem.unn.runnet.ru
Summary.
The Chemical Department, Nizhnii Novgorod University, Gagarin Ave., 23. Nizhnii Novgorod 603600 Russia E-mail: zelen@ichem.unn.runnet.ru Summary. The mechanism of polymer formation in the course of
organic azides photolysis in the crystalline state was studied. It was shown that the
mechanism consisted of (i) formation of nitrenes, (ii) attack of the nitrenes onto the
nitreneous atoms of azido groups with subsequent elimination of molecular nitrogen from
the azide molecules having been attacked, (iii) in the case of aromatic diazide that had
conjugation of aromatic system with both azido groups the second (undecomposed) azido
group could decompose to form the second nitreneous center without absorption of an
additional u.v. photon. The latter may be thermally activated. The role of packing of
azides molecules in the crystal has been revealed. Keywords: organic azides, photolysis, crystalline state, the
reaction mechanism, nitrenes. The organic azides decompose under u.
Reiser and coworkers [3] is seemed to be the first who have described the mechanism of the polymer formation. They proposed that the polymer molecules
were formed as the result of singlet nitrene, RNS, insertion into C-H bond of phenyl rings.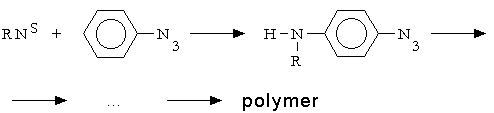
Almost simultaneously Bö ssler and Schultz [4] reported that the polymers formed from aromatic azides under u.v. irradiation
were azopolymers. Unfortunately, they did not describe the reaction mechanism.The formation of plasma resistant azopolymers has been postulated also in [9] and in japanese works [10,11]:
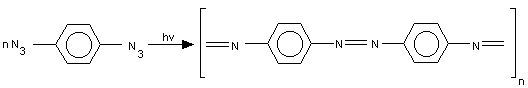
and in [12]
Another japanese research group [13] to interpret photoelectron spectra of polymer being formed under u.v. irradiation of phenylazide freezed onto a gold substrate at 77 K return
ed to the idea proposed by Reiser and Leyshon (reaction 1). These workers wrote down the polymerization mechanism as on the following scheme.
The attempt to deta
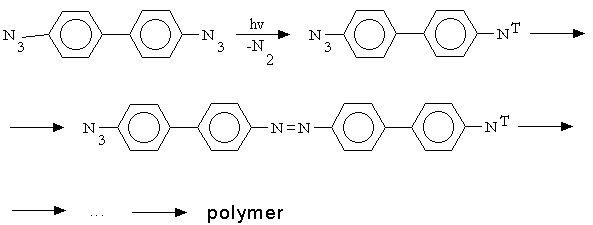
ilize the mechanism of the azopolymer formation under u.v. irradiation of aromatic azides in the crystalline state was undertaken in [14-16]. At the first step of the mechanism proposed in [14] there has place an azide decomposition to form a nitrene in the triplet (ground) and the singlet (excited) state.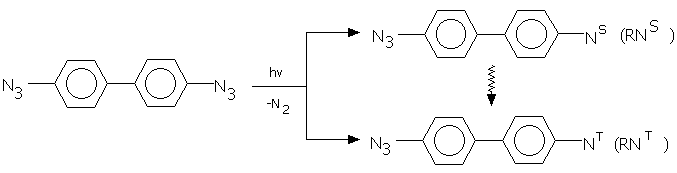
The nitrenes formed
were able to react with adjacent azides molecule, with the reaction having been carried out without an additional u.v. light absorption.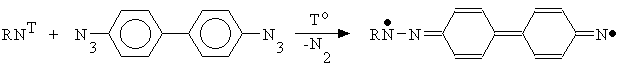
In the next step the biradical formed could react with another adjacent azide molecule and so on up to the azopolymer formation.
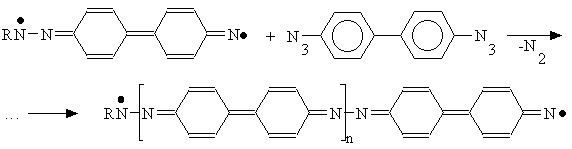
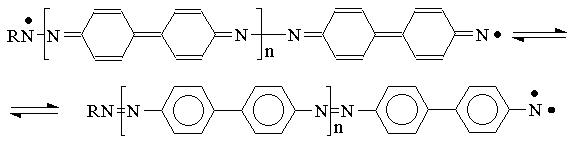
The authors of [15] has shown that the mechanism had to be implemented with an H atom abstraction reaction in the case of vacuum deposited layers made of 4,4’-diazidediphenyl (that considered [15] as being amorphous):
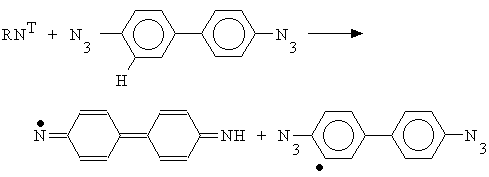
T
he recombination of the nitrene and an hydrocarbon radical occurred as following: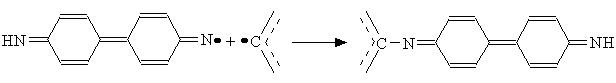
Recently the workers [8] from Holland have proposed the polymer formation mechanism consisting of aromatic azide photopolymerization with didehydroazepine derivatives participation as intermediates.
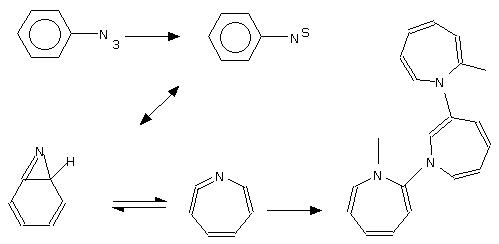
Thus our brief review has shown that either the chemical nature of the polymeric product being formed under an aromatic azide photolysis in the crystalline state or the mechanism of the polymer formation are far away to be
Thus our brief review has shown that either the chemical nature of the polymeric product being formed under an aromatic azide photolysis in the crystalline state or the mechanism of the polymer formation are far away to be
Thus our brief review has shown that either the chemical nature of the polymeric product being formed under an aromatic azide photolysis in the crystalline state or the mechanism of the polymer formation are far away to be
Thus our brief review has shown that either the chemical nature of the polymeric product being formed under an aromatic azide photolysis in the crystalline state or the mechanism of the polymer formation are far away to be established.
.To determine the chemical nature of polymer product formed in the aromatic azide photolysis in the crystalline state we used thin layer chromatography (TLC), IR spectroscopy, and u.v. spectroscopy. In addition we simulated aromatic azide crystal packings with atom-atom potential procedure and with envelop trajectory method [5,6] and
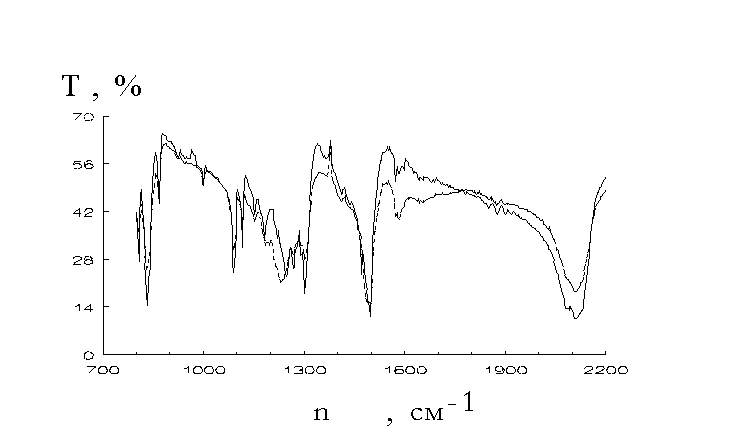
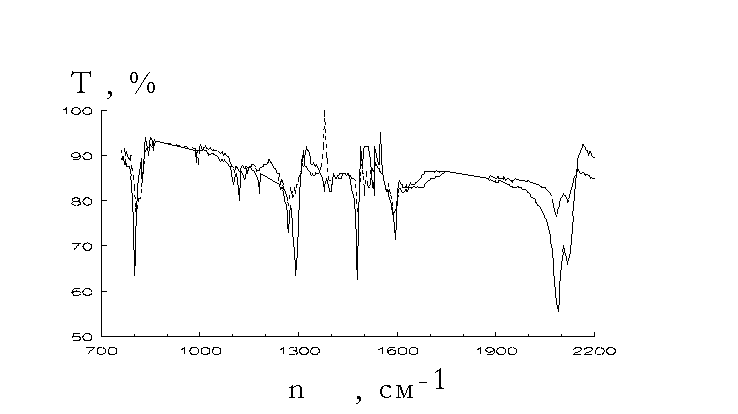
did quantum chemistry calculations of some reaction paths of the mechanism presented by reactions 5-7 [17,18].IR spectra investigations could give an important information on the structure of the polymers formed. Fig.1-3 show
s changes of the IR spectra under the photolysis of aromatic azides. They were recorded with SPECORD 75 IR spectrophotometer. Mixtures of the irradiated azide powders and KBr have been pressed into tablets, the latter being used for IR spectra recordings.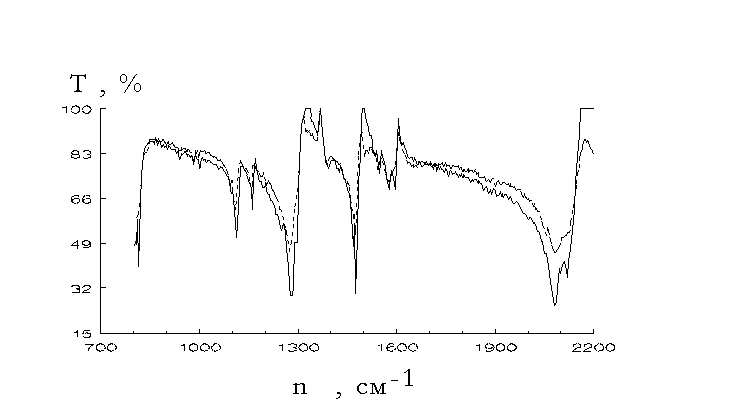
A
fter u.v. irradiation of aromatic azides there had place the lowered N3 group valence vibrations at near 2100 cm-1 and 1300 cm-1 and lowered absorptions at 1600-1480 cm-1 and 860-800 cm-1. The changes could be consequences of the lowered p -electron density delocalization in the aromatic rings (for instance, caused by quinonoid structure formation) [22]. In addition we observed increased absorptions at 1650-1590 cm-1 and 1340-1250 cm-1. They could be caused by some reasons such as quinonoid structure formation, azocompound involvement (as it was shown [4,23] azocompounds absorb in the region), C - N and N - H bond presence. Looking through the spectra shown in Fig.1-3 it seems to be necessary to note that there has no place an increase of absorption at 850-690 cm-1 that they assigned [8] to polymeric 1,2-azepines. There seems to be no reasons to assert that there has place polyazepine formation.It was
shown [14] that the amine yield was low (not more than 0.01% in the case of 4,4’-diazidediphenyl [14]). It seems to be unlikely that absorption at 1650-1590 cm-1 could be assigned to NH deformation vibrations. At the other hand reduction of the azopolymer with SnCl2 in concentrated acetic acid gave primary amines. They were identified with the Erlich reaction [19]. The possibility of such identification was proved by comparison of u.v. spectra recorded in the cases of the reduction product obtained from 4,4’-diazidediphenyl photolysis and 4,4’-diaminediphenyl treatment with Erlich reagent (see Fig.4).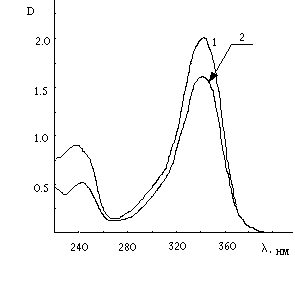
Fig.4 U.V. spectra of diaminediphenyl (1) and the product of reduction of
the 4,4’-diazidediphenyl (2) photolysis polymeric product by means of SuCl2 in conc. acetic acid treated by Erlich reagent.Thus we have had to conclude that IR spectroscopy together with TLC techniques g
ave us an opportunity to assert that the polymeric products were azopolymers.E
arlier it was pointed out [5,6] that the packing of azide molecules in photoreactive crystals has a great influence upon the azopolymer output. The intermolecular electronic density transfer such as in the reaction 6 is possible only in the case when interatomic distance between “nitreneous” nitrogen atoms of N3 groups of the azide molecules being adjacent in the crystal packing is close to a sum of van-der-Vaalse radii of nitrogen atoms (0.30 nm according to [20]). The packing of 4,4’-diazidediphenyl molecules in crystals seems to be the closest one [5,6]. It has stacked layers; the layers being dense packed. It is the case where we have to observe the maximum yield of azopolymer [5,14] and the maximum sensitivity to u.v. irradiation [6]. The yields of substances formed with reaction 9 were [5] ultimately low and not more than 0.01%. The importance of azide packing has been supported with the following. Although 4,4’-diazidediphenyl and 4,4”-diazide-p.-terphenyl have the same type of packing the distance between “nitreneous” nitrogens of azido groups belonging to the adjacent azide molecules is 0.32 and 0.36 nm, correspondingly. The latter made the contribution of the reaction 9 much greater in the case of 4,4”-diazide-p.-terphenyl.In addition to the distance between the “nitreneous” nitrogen atoms of azido groups of molecules that are adjacent in the crystal packing there has place a great influence of energy that being necessary to form the transitional state structure for the reaction between azide and nitrene molecules.
T
he movements of molecules in the dense layer of azide molecules that were necessary to produce the transitional state for the reaction to have place are shown on Fig.5.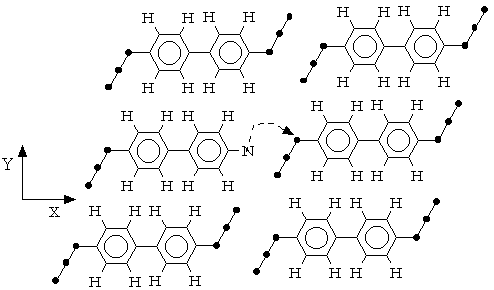
Fig.5 Reaction mechanism of polymer formation in solid state photolysis of aromatic azide.
F
igure 6 shows that the movement of azide molecule along its long axis (X direction) is more energetically preferable compared to the movement of the molecule along its short axis (Y direction) or the interlayer movement (in the directions being perpendicular to the X-Y plane).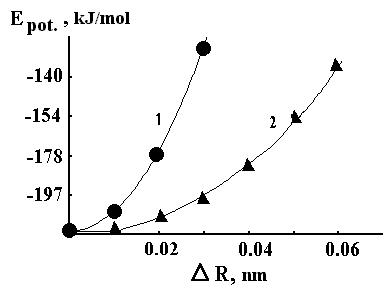
Fig.6 Changes in energy of azide molecule in crystal environment during its movement from equilibrium position: 1 - out of plane and along axis Y (that coincides with long axis of the azide molecule); 2 - along axis X that coincides with shoot axis of the azide molecule).
The next argument proving the possibility of azide decomposition result
ed because of intermolecular electron density transfer between nitrene and azide molecules was obtained by means of quantum chemical calculations. The MNDO procedure was used to model HNT + HN3 ® HNNHT + N2 reaction.Fig.7a show
s the changing of aside group charge during movement of HNT towards “nitreneous” atom of HN3. Fig.7b shows energy changes as a result of the movement described above. It is evident from Fig.7a and 7b that there has place “breaking of the monotonous changing” of the charge caused by intermolecular electron density transfer. The range of intermolecular distances where the said “breaking” having place corresponds to the range where maximum of the energy barrier separating the starting molecules (HNT and HN3) and products having place. An electron transfer into N3 group has had to cause azide molecule decomposition accompanied with N2 elimination [21].(a)
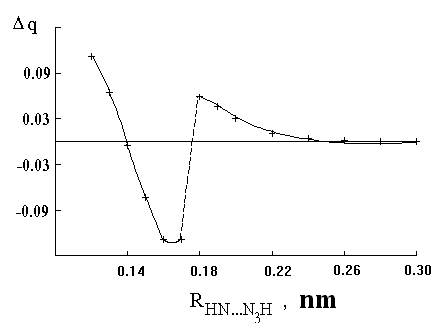
(b)
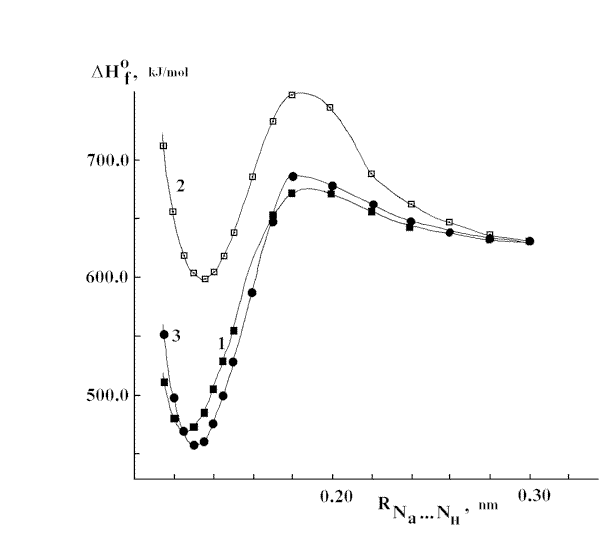
Fig.7 (a) Changes of net charge (compared to the isolated molecule) of N3 group of HN3 molecule during the movement of NH towards
the “nitreneous” nitrogen of N3 group in HN3 molecule; (b) formation enthalpies of {HN3 +HN}T system as a function of interatomic distance between nitrogen of NH molecule and “nitreneous” nitrogen of HN3 molecule.Acknowledgment. We thank the Interdepartmental Scientifical and Technical Program "Chemistry" ("Photochemistry" division) (Russian Gov.Com.) for financial support of the research.
References
.1. Horner L., Christmann A., Gross A.//Chem.Ber.1963.Bd.96.S.388.
2. Lwowski W., Scheiffell E.//J.Am.Chem.Soc.1965.V.87.P.4359.
3. Reiser A., Leyshon L.J.// J.Am.Chem.Soc.1971.V.93.No.16.P.4051.
4. B?ssler H., Schultz R.C.//Makromol.Chem.1972.Bd.158.S.113.
5. Zelentsov S.V., Oleinik A.V., Karjakina L.N.// 4th Conf. “Non-silver and unusial photographic processes". Abstr.Book Vol.2.
Tchernogolovka.1984.P.43 (in Russ.)
6. Oleinik A.V., Zelentsov S.V. // Proc. of Nizhnii Novgorod Univ. 1994. P.189. (in Russ.)
7. Moreau W.M. Microdevices. Physics and Fabrication Technologies. Plenum Press. New York. 1988.
8. Meijer E.W., Nijhuis S., van Vroonhoven F.C.B.M.//J.Am.Chem.Soc. 1988. V.110.No.21.P.7209.
9. Cox R., Clecak N., Moreau W.M.//Polym.Eng.Sci.1974.V.14.P.491.
10. Yabe A., Niino H., Moriyama H., Ouchi A.//Chem.Funct.Dyes: 1st Int. Symp.Tolego.1989.P.206.
11. Yabe A., Ouchi A., Moriyama H., Masuda E.// US Pat. No.4875986. Cl.204/157.6. Publ.24.10.89.
12. Kobayashi T., Miura A., Ohtani O., Yamaoka T.//Conf. Laser and Electro-Opt., OSA/IEEE Div.Techn.Pap.Washington.1986.P.36
13. Harada Y., Takahashi T., Mutai K. //J.Chem.Phys.1981.V.75.No.4. P.2014.
14. Zelentsov S.V., Oleinik A.V., Karjakina L.N.//High Energy Chemistry. 1986.V.20.No.4.P.339.
15. Karjakina L.N., Zidkova E.M., Janin A.M., Oleinik A.V.//High Energy Chemistry. 1987.V.21.No.4.P.348.
16. Zelentsov S.V., Oleinik A.V. //Conf. “Radical Polymerization”. Gorkii. Abstr.Book.1989.P.238. (in Russ.)
17. Zelentsov S.V., Kokorev V.N., Oleinik A.V. // 4th Conf. on Carbene Chemistry. Moscow.Nauka.1987.P.52. (in Russ.)
18. Kokorev V.N., Zelentsov S.V., Oleinik A.V. // 5th Conf. on Carbene Chemistry. Moscow.Nauka.1992.P.162. (in Russ.)
19. Laboratory Handbook of Chromatographic and Applied Methods/ Ed. by O.Mikes.Ellis Horwood Ltd. Publishers.New York e.a. 1979.
20. Zefirov U.V., Zorkii P.M. // J. Struct.Chem. (Russia).1974.V.15.No.1. P.118.
21. Kononenko L., Urre T.A., Dmitrieva V.N., Efros L.C., Bezhuglii V.D. // J. Gen.Chem. (Russia) 1970.V.40.No.6.P.1359.
22. Smith A.L. Applied Infrared Spectroscopy. Fundermentals, Technics, and Analitical Problem-Solving. John Wiley and Sons Publ.
New York e.a. 1979.
23. Dyer J.R. Applications of Absorption Spectroscopy of Organic Compounds. Prentice-Hall Inc. New-York e.a.