| Papers and Posters | Site Home Page |
The organic azides photooxidation: brief review
Sergey V. Zelentsov, Nadezda V. Zelentsova, Alexcej B. Zheslov
Chemical department,
Nizhnii Novgorod State University,
23, Gagarin Ave., Nizhnii Novgorod, 603600, Russia
Tel.: +7 (831 2) 657 227
Fax: +7 (831 2) 658 592
E-mail: zelen@ichem.unn.runnet.ru
Summary
The mechanisms of organic azide photolysis in presence of oxygen are briefly reviewed.
A new reaction scheme of the process is proposed. Adducts of nitrene with O2 in the triplet and singlet states are considered to be the key intermediates. There are three main paths of the photooxidation: (i) non-nitrenious one proposed by Abramovitch et. al., (ii) an abstraction of hydrogen atoms from a solvent by the triplet adduct to give a radical N-hydroperoxide followed with interactions of the radicals involved and decomposition of the N-hydroperoxide to produce nitrosocompound and hydroxyl radical, and (iii) the singlet adduct reactions to produce nitrocompound. In addition to the main channels the triplet adduct can oxidize nitrosocompound to nitrocompound. The rate determining step of the process is the singlet-triplet azide intercrossing.
Photooxidation of organic azides is one of the most poorly investigated reactions of the azide photochemistry. There are only scarce works on the subject. Data and interpretations contained in them are not always concrete and unanimous.
Abramovitch and co-workers [1,2] were the first who began to study the reaction. They found out that the main products were nitrocompounds, azoxycompounds and nitrosocompounds, the latter considered to be the primary stable products. The authors proposed the following "non-nitrenions" mechanism.
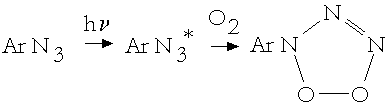 (1)
(1)
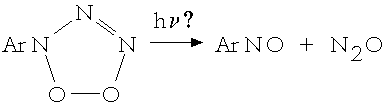 (2)
(2)
![]() (3)
(3)
More carefully the photooxidation products were studied by Lee Go and Waddell [3]. After prolonged photooxidation of phenylazide in acetonitrile azobenzene, nitrobenzene, and nitrosobenzene were isolated. In low concentrated solution the reaction gave only nitrosocompound.
Almost at the same time Brinen and Singh [4] demonstrated that there were two key intermediates in the photooxidation reaction: paramagnetic adduct,
RNOOT, and the diamagnetic one, RNOOS. The intermediates were observed at 77 K after irradiation of frozen azide solutions saturated with O2.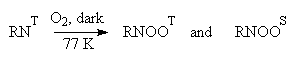 (4)
(4)
Although there are no unanimous results on the spin nature of the ground state of RNOO, it seems [4,5] to be triplet. The conclusion is in contradiction to one made by Pritchina and Gritsan [6] who found out that the ground state of
para-NH2C6H4NOO was singlet.Brinen and Singh [4] proposed also that particles
were formed in the course of the photooxidation. Their decomposition gave nitroso-compounds and nitrocompounds.
The path with RNOOS participation was investigated by Pritchina and Gritsan [6-8]. The authors stated that para-nitrophenylnitrene reacted with O2 to produce para-C6H4N=O+-O- being a diamagnetic adduct. In toluene-tetrahydrofurane (1:1) matrix at 77 K the adduct could exist in two different forms having l max at 495 and 588 nm. They could be transformed each other under u.v. irradiation having wavelength of 436 and 577 nm.
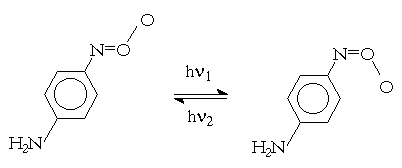 (5)
(5)
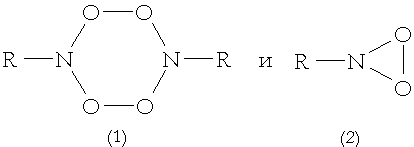
RNOOS particles died due to the second order kinetics low to produce tetraoxide 1 [4,6], and the rate constant of the reaction was (1.4± 0.4)· 109 M-1· s-1 (in toluene) and (0.7± 0.2)· 109 M-1· s-1 (in n.-hexane) [8]. The u.v. spectra of the compound had absorption with maximum at l =336 nm (in methylcyclohexane). The life-time of the tetraoxide 1 was about 1 s at 300 K. As a result of parallel reactions the tetraoxide could give nitrocompound and nitrosocompound, and polarity or polarizability of solvent increase (as a result of Et3N addition) enlarged the nitrosocompound relative yield.
The next stride was made by Japanese authors [9,10]. They established three important facts. The first one was that RNOOT particle could play the role of an electrophilic oxygen atom carrier and oxidize benzene to phenol, Ph2S to Ph2SO, ArNO to ArNO2 etc. For instance, photooxidation of phenylazide in acetonitrile containing 10% of benzene gave the phenol yield of about 15% [9].
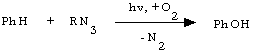 (6)
(6)
The second observation of the authors was that mixed nitrocompounds, R16O18O, were formed in the course of photooxidation with 16O2 enriched by 18O2.They proposed the following mechanism to explain the result.
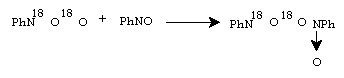 (
(
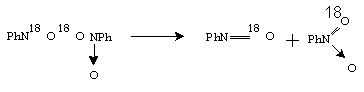 (
(
Lately these authors [10] determined relative reactivities of RNOOT (R = para-MeOC6H4) towards hydrooxidation of hydrogen atoms bound to primary, secondary, and tertiary carbon atoms. The said values were 1:8:49 [10]. These ratios were close to ones typical for · OH relative reactivities [10].
Studying of the 3- and 4-nitrophenylazide photooxidation gave Liang and Schuster opportunity to state [11] that the phooxidation products yields depended upon intensity of u.v. irradiation. Low intensity irradiation of the azides solutions saturated with O2 gave 20% of azocompounds, 11% of nitrosocompounds, 12% of nitrocompounds and 28% of azoxycompounds. High intensity irradiation gave 97% of azocompounds and 3% of nitrocompounds. In addition when low intensity irradiating was used the yields of the products seemed to depend upon conversion of azides, an increase of the latter produced a decrease of the nitrosocompound yield and an increase of the azoxycompound yield [11].
The authors made another important observation that nitrosocompounds addition to the photooxidizing azides prohibited the oxygen containing products formation [11].
The intriguing feature of the aromatic nitrenes is their lowered oxidation rate constants (see table 1). The values are 104-105 times less than the oxidation rates of the triplet carbenes or of the simplest nitrene, NH.
Table 1. Nitrene oxidation rate constants.
| nitrene | rate constant, M-1× s-1 | literature | ||
| NH 2 ,4,6-tribromophenylnitrene4- nitro-1-nitrenobenzenephenylnitrene 4- aminophenylnitrene |
|
11 10 10 12 8 |
There is no satisfying explanation of the phenomena. The lower reactivity of phenylnitrene compared to phenylcarbene may be accounted for changes of electron density distribution in the molecules [14]. Comparison of the electron interactions that have place during oxygen molecule approaching to a nitrene and a carbene can reveal existence of the additional interelectron repulsion in the nitrene, the latter being absent in the carbene [14]. Although the interaction does decrease the relative reactivities of nitrenes it is hardly able to lower them for 105 times. The latter conclusion is in accordance with the rate constant of the triplet para-nitrophenylnitrene recombination of 1.0· 109 M-1s-1 [4,15] that is only 10 times less than the diffusion limit. It is relevant to note that the only reaction that has the ratio of the arylnitrene reactivity to the arylcarbene one close to the photooxidation is the intercrossing of the singlet nitrene to the triplet nitrene (that values are 105.4-6.2 s-1 and 109-10 s-1 for phenylnitrene and phenylcarbene, respectively [16]).
The said observation outlines importance of the nitrene spin state when this intermediate reacts with O2. Abramovitch and co-workers [1,2] were able to establish that the nitrocompound yield was 5 times greater in the case of photochemically generated nitrenes than thermally generated ones. They also stated that the ratio of the nitrocom-pound yields produced in the course of ferrocenyl azide and phenyl azide photooxidation was 4:1 [1,2]. Our preliminary results [17] shows that the triplet photosensitizers increase the nitroso-compound yield, nitrocompound yield being approximately unchangeable (up not more than 25% conversion of azide). The photooxidation in the solvents that are able to stabilize the singlet particles gives the nitrocompound yield increase.
Earlier the effect of the nitrene singlet state stabilization in CHCl3 on the photo-oxidation product yields was observed by Mamaev and co-workers [18]. They found that the nitrocompound yield increased from 35% in ethanol to 63% in CHCl3 and from 30% in ethanol to 65% in CHCl3 in the cases of the 2-azidophenylpyrimidine and 4-chloro-phenylpyrimidine photooxidation, correspondingly.
Thus the triplet nitrene seems to participate in the path that considers the nitrosocompound as the primary product. On the other hand the singlet stabilization of the intermediates needs to form nitrocompound. The intermediates considered to be RNOOS, in as much as recently Albini et al showed [19] that the singlet nitrene did not react with oxygen to form the oxygen containing photooxidation products.
The organic azides photooxidation reaction was investigated not only in solvents but in the solid state polymer matrices and on the surface of crystals [20-31]. It was shown [20-22] that very stable (at room temperatures) radicals having the non-asymmetrical ESR spectrum shown on fig.1 were observed in the reactions. One of the possible inter-pretation was to consider them as superposition of ESR spectrum of the macromolecular carbon-centered radical and of the peroxide radical [20-22]. To reveal ways of the peroxide radical formation the authors proposed [22] that along with interconversion of RNOOS to nitrocompound there had place an hydrogen atom abstraction from polymers to produce peroxide and macromolecular radicals.
![]() (9)
(9)
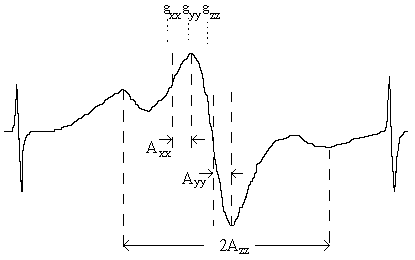
Fig.1 ESR spectrum of the organic azides photooxidation paramagnetic products stabilized in polymer matrices [23,27]
As it was noted [22] the super-stabilization of the said radical pair in rigid matrices was completely kinetical one being one of the results of the reactions:
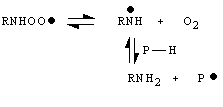 (
(
![]()
On the basis of reactions 9-11 the vast array of experimental results was explained. The array consisted of the following data: (i) inhibition of photocrosslinking of polymer with organic diazides [23]; (ii) disappearance of the ESR spectra of photooxidized azides after dissolving of the azzides in organic solvents [24], after treatment of them with iodine vapour, NH3, or NO [24], after action of surface-acoustic waves upon the films [25]; (iii) packing density of the polymer matrix used has a tremendous influence on the yield of the radical centers after u.v. irradiation of the systems in O2 presence; the effect depending of the method of the matrix preparation (i.e. drying characteristics, type of the solvent used, storage time of the polymer solutions that contained organic diazides etc [26].
The chemical nature of the stable radicals observed after photooxidation of organic azides in polymer matrices was carefully studied in [27,28]. The authors used the ESR spectra model that considered the spectra having anisotropic both g-factor and super fine coupling tensor [27]. Table 2 shows some of the results.
Table 2. Typical ESR spectra parameters of organic azides observed in the course of their u.v. irradiation in rigid matrices [27].
|
gzz | gyy | gzz | Azz, G | Ayy, G | Axx, G | ||
| 4,4'-diazidodi-phenyl in poly-sterene matrices | 2.0027± 0.0004 | 2.0059± 0.0005 | 2.0086± 0.0007 | 25.7± 0.3 | 5.0± 0.2 | 4.5± 0.2 | ||
| 4,4'-diazidodi-phenyl on crys-tal surfaces | 2.0028± 0.0005 | 2.0055± 0.0004 | 2.0084± 0.0006 | 24.7± 0.3 | 4.6± 0.3 | 4.5± 0.4 | ||
| Ph2NO· *) | 2.0022 | 2.0056 | 2.0092 | 23.8 | 3.6 | 1.9 | ||
| 2,2,6,6-tetrame-thyl-4-oxopype-ridine-1-oxide in tetramethyl-cyclobutadione solution**) | 2.0027 | 2.0061 | 2.0089 | 33.4 | 4.8 | 4.8 |
*)
- taken from [11];**)
- taken from [13].Comparison of the paramagnetic parameters of the photooxidation products and the iminoxyl radicals known from literature gives us an opportunity to state [27] that the paramagnetic photooxidation products in the solid state seem to be iminoxyl radicals. Using the saturation of ESR spectra the authors [28] were able to prove that the iminoxyl radical center was bound to a polymer main chain or localized on the surface of the organic azide crystal, the corresponding spin-lattice relaxation times were in the interval from 4.00· 10-6 s to 21.20· 10-6 s.
Recently chemical nature of the final diamagnetic products of the photooxidation was studied [31]. It was shown that the photooxidation products composition was different when the reaction had place in the liquid and in the solid state (see table 3). The main products of the reaction occurred in the solid state polymer films (polystyrene) were nitrosocompound and azocompound, and their yields were approximately the same for all azides studied, the formation of nitrocompound was suppressed. In benzene the main products of the reaction were azocompound and nitrocompound, the yield of nitrosocompound was 10 times less. The results obtained are in accordance with the suggestion [32,33] that nitrosocompound may be formed as a result of the following reaction
![]()
Table 3.
The organic azides photooxidation products yields (w.%) in polymer films and in benzene.| Products | Polymer in the solid state | Benzene | ||
| 1,4-diazido-benzene | 4,4'-diazidobi-phenyl | 4,4'-diazidobi-phenyl ester | 4,4'-diazido-biphenyl | |
| Nitrosocompound | 45.0 | 52.5 | 48.6 | 5.6 |
| Azoxycompound | 4.1 | 4.0 | 3.5 | - |
| Nitrocompound | - | - | - | 13.7 |
| Azocompound | 18.2 | 15.0 | 15.0 | 28.0 |
| conversion, % | 25.0 | 25.6 | 28.0 | 25.0 |
Nitrocompounds may be formed in the course of the secondary reactions, for instance, due to nitrosocompound oxidation with RNOOT particles [9,31]. The latter reaction following to the second order kinetics could be suppressed in the media of low reagents mobilities.
Interesting results were obtained with the method of competing reactions [34,35]. In the work [35] it was studied photocrosslinking of polymers with organic diazides in polymers in presence of oxygen. Monoazides that could be photooxidized but was not able to form additional crosslinks were added into the reaction system. It was found [35] that the monoazide addition gave an increase of the photooxidation effectiveness; the latter increased with the azide molecule size increasing. To explain the results the following suggestions were made: (i) oxygen could be absorbed reversibly (and then the photooxidation product yield determined by competition of the diazide and the monoazide photooxidations; (ii) the photooxidation products (or the intermediates involved) could serve as "trapping agents" of nitrenes.
More detailed studying of the azide molecule size influence upon the effectiveness of the photooxidation was performed in work [36]. It was stated that the paramagnetic product yield increased when the size of the azide molecule to photooxidize was increased. The dependence had exponential behavior. There existed the critical size from which the asymmetry of the ESR spectra appeared.
The results described in the previous part of present paper can be rationalized on the basis of the following "general" photooxidation scheme.
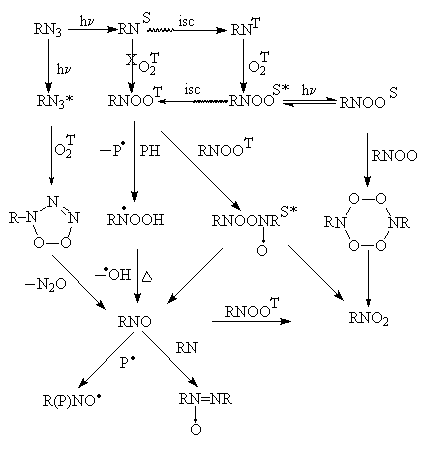
On the first step azide can be excited with or without production of singlet nitrene. The latter possibility gives rise to the so called "non-nitreneous mechanism" that was proposed by Abramovitch and co-workers [1,2] and the existence of which was "felt" by Liang and Schuster [11]. The singlet nitrene can intercross to the triplet (ground) state. This very slow process is the rate determining step of the photooxidation [16]. Usage of the triplet photosensitizes can avoid the slow step and increases the photooxidation effectiveness.
The triplet nitrene can react with oxygen to produce the nitrene-oxygen adduct in the excited singlet state. The latter can either produce the adduct in the triplet state or lose excessive energy. The triplet adduct either reacts with solvent molecules to produce radical N-hydroperoxide that can in its turn dissociate to give nitrosocompound and hydroxyl radical. There are at least to two evidences to support the existence of the path. The first one is relative reactivities of hydroxyl displacement in benzene derivatives that are close to the values typical for the hydroxyl radical attack [10]. The second one is that other ways described include the bimolecular processes such as RNOOT autooxidation or destruction of hypothetical tetraoxide (that also can be formed only as a result of the bimolecular reaction of RNOOS dimerization) and they are to be suppressed in the case of low reagents mobilities. So the nitrosocompound yield increases in the solid state [31] compared to solution.
The singlet adduct can either dimerize to give tetraoxide and then dissociate to produce nitrosocompound and nitrocompound or rearrange (photochemically?) to form nitrocompound. The existence of the latter path is supported by the results obtained when photooxidation of the organic azides has place in the solvents stabilizing the singlet states of molecules (in the case an enhanced yield of nitrocompound) is observed. Having in mind that the singlet nitrene is oxidized very slowly [19] it is possible to conclude that the stabilizing solvents stabilize the singlet nitrene-oxygen adduct. The results suggest that nitro-compound can be formed not only in the ways with the triplet adduct participation but in an independent path.
Being the primary product nitrosocompound can produce a great deal of other products such as iminoxyl radicals, azoxycompounds, nitrocompounds etc.
The scheme does not show "non-oxidation" reactions of triplet and singlet nitrenes such as dimerization, hydrogen abstraction, insertion in various bonds etc. Of course the latter reactions complicates the photooxidation. It is enough only to mention one fact. Poe et al found that when the singlet nitrene is stabilized enough (as in the case of C6F5N) oxygen can influence only upon relative yields of various insertion products but does not form any oxygen-containing product [37].
In addition oxygen can be trapped by aminoradicals RN· H (derived from triplet nitrene hydrogen abstraction reactions) due to reaction
![]() (13)
(13)
To conclude we should to note the following. The organic azide photooxidation reactions are very interesting and promising field to investigate. We tried to give brief review of the results obtained in the field and to rationalize them with one of possible schemes. It needs careful testing and further improvements.
Acknowledgment. We thank the Interdepartmental Scientifical and Technical Program "Chemistry" ("Photochemistry" devision) (Russian Gov.Com.) for financial support of the research.
References
1.
R.A.Abramovitch, C.I.Azogu, and R.G.Sutterland // J.Chem.Soc., Chem. Commun. 1971. No.3. P.134.2.
R.A.Abramovitch and S.R.Shalland halland // J.Chem.Soc., Chem.Commun.1972. No.16. P.964.3.
C.Lee Go and W.H.Waddell Waddell // J.Org. Chem. 1983.V.48.No.17.P.2897.4.
J.S.Brinen and B.Singh Singh // J.Am.Chem.Soc.1971.V.93.No.24. P.6623.5. P.C.Hiberty and G.Ohanessian // J.Am.Chem.Soc. 1982. V.104. No.1. P.66.
6
. E.A.Pritchina and N.P.Gritsan // J.Photochem. and Photobiol.A.:Chemistry. 1988. V.43. P.165.7
. E.A.Pritchina, N.P.Gritsan, and N.M.Bazhin and N.M.Bazhin and N.M.Bazhin // Izv.Akad.Nauk SSSR.Ser Chem. 1986. No.8. P.1749.8
. E.A.Pritchina and N.P.Gritsan // Kinetics and Catalisis. 1987. V.28. No.5. P.1044 (in Russ.).9
. Yasuhiro Suwaki, Shinji Ishikawa, Yasuhiro Suwaki, Shinji Ishikawa, Yasuhiro Suwaki, Shinji Ishikawa, Yasuhiro Suwaki, Shinji Ishikawa, and Hiizu Iwamura Hiizu Iwamura // J.Am.Chem.Soc. 1987. V.109. No.2. P.584.10. Shinji Ishikawa, Takaynki Najima, and Yasuhiko Sawaki // J.Chem.Soc. Perkin.Trans. 2. 1996. No.1. P.127
1
1. T.-L.Liang and G.B.Schuster Schuster // J.Am.Chem. Soc.1987. V.109. No.25. P.7803.1
2. F.Rohrer and F.Stuhl //J.Chem.Phys. 1987. V.86. No.1. P.226.13. E.
E.Leyva, M.S.Platz, G.Persy, and J.WirtzWirtz // J.Am.Chem.Soc.1986. V.108. No.13. P.3783.1
4. H.Zhai and M.S.Platz //J.Phys.Org.Chem. 1996. V.10. No.1. P.22.15.A.V.El'tsov, I.G.Batekha, G.Bekker, H.Betyer, V.S.Kuznetsov, O.P.Studzinski, V.E.Holmogorov, K.Schilder, and T.A.Urre Photochemical processes in layers. Chemia. Leningrad.1978.P.118 (in Russ).
16
. N.P.Gritsan, T.Yuzawa, and M.S.Platz //J.Am.Chem.Soc. 1997. V.119. No.21. P.5059.17. A.B.Zhezlov
A.B.Zhezlov, O.A.Tachaeva, S.V.Zelentsov, and A.V.Oleinik //Proc of the Nizhnii Novgorod Univ.Chemistry.1998. No.1.P.89. (in Russ.)18. V.P.Mamaev, T.A.Andreeva, V.P.Krivopalov, and V.I.Eroschkin // Izv.Akad.Nauk SSSR. Ser.Chem.1986. No.4.P.955
19. A.Albini, G.Bettinetti, and G.Minoli // J.Am.Chem.Soc.1997. V.119. No.31. P.7308.
20. V.M.Treushnikov, L.L.Pomerantseva, N.V.Frolova, and A.V.Oleinik // J.Appl. Specrtoscopy (Russia). 1979. V.30. No.5. P.929 (in Russ.).
21. A.M.Yanin, L.N.Kariakina, V.M.Treushnikov, A.V.Oleinik, and E.L.Sorin // High Energy Chem. 1987. V.21. No.2.P.164. (in Russ.).
22. V.M.Treushnikov and N.V.Frolova // Visokomol.Soedin. (High polymers). A. 1983. V.25. No.7. P.1400 (in Russ.).
23. A.V.Oleinik,V.M.Treushnikov, and N.V.Frolova // Z.
Nauchn. i Prikladn. Fotograf. i kinematogr. 1975. V.20. No.5. P.361 [Journal of the Scientific and Applied Photography and Cinematography.- in Russ.].24. V.M.Treushnikov, L.L.Pomerantseva, N.V.Zelentsova, and A.V.Oleinik // Visokomol. Soedin. (High polymers). A. 1983. V.25. No.5. P.327. (in Russ.).
25. D.S.Volgunov, S.A.Yesin, N.V.Zelentsova, S.G.Petrov, and V.M.Treushnikov // Z.
Nauchn. i Prikladn.Fotograf. i kinematogr. 1987. V.32. No.2. P.87 [Journal of the Scientific and Applied Photography and Cinematography.- Russ.].26. N.V.Zelentsova, V.M.Treushnikov, and A.V.Oleinik // Visokomol.Soedin. (High polymers). B. 1984. V.26. No.1. P.32. (in Russ.).
27. S.V.Zelentsov, A.V.Eryutov, A.A.Ezhevskii, and A.V.Oleinik // High Energy Chem. 1997. V.31. P.193. (Engl.Ed.).
28. S.V.Zelentsov, E.A.Bykova, A.A.Ezhevskii, and A.V.Oleinik // High Energy Chem. 1997. V.31. P.397. (Engl.Ed.).
29. A.N.Kuznetsov Metod spinovogo zonda. (Method of spin probes). Moscow. Nauka. 1976. [in Russ.].
30. Spin Labeling. Theory and Applications / Ed. by L.J.Berliner. Academic Press. New York-San Francisco-London. 1976.
31. A.B.Zhezlov, S.V.Zelentsov, and A.V.Oleinik // High Energy Chem. 1999. V.33. No.2 .P.122. (Engl.Ed.).
32. S.V.Zelentsov // Proc.of the 6th all-union conf. on photochemistry. Part 1. Novosibirsk. I.Ch.C.C.SP AS USSR. 1989. P.168 (in Russ.).
33. S.V.Zelentsov // Proc. of the 4th conference on carbenes. Moscow. Nauka 1987. P.49 (in Russ.).
34. S.V.Zelentsov and A.V.Oleinik // Z.
Nauchn. i Prikladn. Fotograf. i kinematogr. 1983. V.28. No.4. P.298 [Journal of the Scientific and Applied Photography and Cinematography.- in Russ.].35. S.V.Zelentsov, N.V.Zelentsova, V.M.Treushnikov, and A.V.Oleinik // Z.
Nauchn. i Prikladn. Fotograf. i kinematogr. 1983. V.28. No.5. P.359 [Journal of the Scientific and Applied Photography and Cinematography.- in Russ.].36. S.V.Zelentsov, N.V.Zelentsova, and A.V.Oleinik // High Energy Chem. 1989. V.23. No.4 .P.358. (Russ.Ed.).
37. R.Poe, K.Schnapp, M.J.T.Young, J.Grayzar, and M.S.Platz //J. Am. Chem. Soc. 1992. V.114. No.13. P.5054.