| Papers and Posters | Site Home Page |
Bilirubin- and light induced cell death in a murine lymphoma cell line
Terje Christensen1,2, Ellen B. Roll1,2, Alicja Jaworska1 and Gunnar Kinn1
1Radiation Medicine Department, Norwegian Radiation Protection Authority and Institutes of Radiology and Pathology, Norwegian National Hospital, University of Oslo, Norway.
2
Institute of Physics, Norwegian University of Science and Technology, Trondheim, Norway.Post address: Norwegian Radiation Protection Authority. P.O.Box 55, N-1332 Østerås, Norway, e-mail: firstname.lastname@nrpa.no, Internet: http://www.nrpa.no
Key words: Phototherapy, newborn, bilirubin, apoptosis, necrosis, blue light, mouse lymphoma cells.
ABSTRACT
Cells from the mouse lymphoma cell line L5178Y-R were exposed to blue light from phototherapy lamps in the presence of solutions of 160 mM bilirubin supplemented with serum albumin. HPLC analysis showed that the bilirubin solution was photooxidised as a function of increasing light dose. The cells were stained with trypan blue to score necrosis, and apoptosis was assayed by the terminal deoxynucleotide assay (TdT) or by studying the nuclear structure in cells stained with propidium iodide.
A rapidly developing apoptosis was observed after light doses killing 60-80 % of the cells as judged from the trypan blue exclusion test. The fraction of apoptotic cells was smaller than the fraction of necrotic cells.
Exposure of the cells to fractions of light at a high dose rate was compared to the effect of a lower dose rate given as a single fraction. No large differences were found, however, there was a tendency of a higher degree of necrosis as well as apoptosis in the cells receiving the light in fractions at a high dose rate.
INTRODUCTION
Phototherapy of neonatal hyperbilirubinemia has been performed for more than thirty years. In most hospitals the light treatment is given as a continuous irradiation only interrupted by short periods of feeding, and the light intensity and colour of the light being under poor control (1). Several methods for improving the effect of phototherapy have been suggested, e.g. the use of more optimal wavelengths (2), more optimal irradiance (3) or fractionation of the light dose. (4)
The choice of the right physical parameters must also take into consideration the possibility that blue light itself or in combination with bilirubin may cause photochemical damage to tissues. The most serious risk is retinal damage if the eyes are exposed without eye shields (5).
It has been shown that bilirubin may induce photosensitising effects on cells in culture (6, 7,8,9).
Cells from several different cell lines may be killed by bilirubin and light, most efficiently when the cells are present in the light field suspended in a solution of bilirubin, but also when added to solutions exposed to high light doses. It is assumed that the toxic effects are caused by hydrogen peroxide and other peroxides (7, 8). The present article deals with the mode of cell death, and it is our intention to further elucidate the possibility that apoptosis or programmed cell death may be induced. Previous observations of damage to DNA, induction of mutations, inhibition of cell multiplication and inhibition of the progression through the cell cycle (10) are of particular interest. Studies of the line 308 mouse epidermal cells indicated that double strand breaks in the DNA were formed, but the morphology of the cells after light irradiation in the presence of bilirubin did not indicate changes typical for late apoptosis (11).
Bilirubin is an antioxidant and has been reported to be a scavenger of peroxyl radicals generated chemically (12,13,14).
L5178Y-R(LY-R)
murine lymphoma cell line was isolated from a methylcholantrene-induced lymphoma in a DBA/2 mouse at the end of the fifties. This cell line, together with its variant (LY-S), is extensively characterised in respect to sensitivity to different types of radiation (ionizing-, UV- and visible-) oxidants and other factors which may influence radiation sensitivity (15, 16, 17). Several studies on DNA damage and repair have been performed and apoptosis has been the subject of studies in the last decade (18, 16). The enzymatic antioxidant system was extensively studied in this cell lines as well (19). Briefly, LY-R cells are relatively sensitive to UV (20) and H2O2, and relatively deficient in H2O2 scavenging enzymes activity, but not in superoxide dismutase.The aims of the present study are:
| To find out whether LY-R cells are killed by an apoptotic mechanism when subjected to light irradiation in the presence of bilirubin. | |
| To determine how fast the apoptotic process proceeds. | |
| To study the dose-response relationship with respect to light. | |
| To study the influence of a light dose split in fractions compared to continuous irradiation. |
MATERIALS AND METHODS
Cell culture
Suspension cultures of mouse lymphoma LY-R cells were cultivated in RPMI 1640 medium
(GIBCO) containing 10 % foetal calf serum. 500.000 cells/ml were dispensed in 25 cm2 tissue culture flasks prior to treatment.
Experimental solution
Bilirubin (BR) (mixed isomers from bovine gall stones, Sigma) was used without further purification. The bilirubin was dissolved in 0.01 or 0.1 N NaOH as a stock solution of 1600 mM and used within four hours. It was filtered through a sterile filter and adjusted to pH 7.2-7.4 with 0.1 N HCl if necessary. The BR stock solution was added to a final concentration of 160 mM BR in solutions of phosphate buffered saline (PBS) with supplements of serum albumin. Two different concentrations of serum albumin were used: Either a dilution of foetal calf serum yielding approximately 50 mM bovine serum albumin (BSA) or a solution of 200 mM human serum albumin (HSA, Sigma, essentially fatty acid free). It should be noted that in the former solution the BR concentration was higher than the concentration of albumin while it can be assumed that most of the BR was bound to HSA in the latter due to the higher molar concentration of HSA compared to BR.
Light irradiation and dosimetry
Irradiation of the cells was performed in flasks on a plexiglass stand placed above two fluorescent tubes (Philips TL20W/52) frequently used in phototherapy. The lamps emit blue light with a spectral distribution peaking at 450 nm. The spectra do not include ultraviolet radiation to any significant extent. The spectrum of the lamps and dose measurements have been published previously (21). The irradiances used in the present experiments were either 5.5 mW/cm2 (I) or half this value (2.7 mW/cm2, I/2) depending on the distance between the lamps and the plexiglass stand. There was a certain variation in irradiance (5-10%) as a function of the position of the flasks in the longitudinal direction of the lamps. To avoid any systematic effect of this variation, the flasks were placed on the stand in random order and the positions of the flasks were exchanged throughout the experiment.
High pressure liquid chromatography
Cell culture flasks (25 cm2) containing 5 ml of bilirubin solution (160 mM BR, 200 mM human serum albumin in phosphate buffered saline (PBS))were placed on top of two fluorescent light tubes as described above (Irradiation section). Mean irradiance along tubes was 5.5 mW/cm2. Samples for HPLC-analysis at a detection wavelength of 450 nm were taken after 0,1, 2.5,5,10 and 15 min. and subsequently every 15 min. for a total period of 120 min. The samples were diluted 1:4 with mobile phase and centrifuged for 5 min. at 5000 rpm. The supernatant was gassed with helium gas prior to HPLC-detection. The HPLC consisted of a Shimadzu LC-10 AT liquid chromatograph, SPD-10 AV spectrophotometric detector, equipped with a Nucleosil 120-5C18 reversed phase column (250x4.6 mm) (Macherey-Nagel AG). Integration was done with Class LC-10 software. The mobile phase consisted of methanol (HPLC-grade, Rathburn), acetic acid (puriss.p.a., Fluka) and destilled water, and pH was adjusted to 7.7 with dioctylamine (for synthesis, Merck). It was continuously gassed with helium gas during analysis, and the flow was set to 1ml/min. The areas under the peaks of the chromatograms were used in calculating the relative concentrations of the bilirubin isomers. The sum of the areas of the 4Z,15Z and the photoisomers (4Z,15E), (4E,15Z), E- and Z-lumirubin was normalised to 1.0. The integrated peaks were corrected for differences in isomer absorption at 450 nm relative to 4Z,15Z. The relative extinction coefficients for 4Z,15Z; 4Z,15E; 4E,15Z; 15Z-lumirubin; 15E-lumirubin at 450 nm are 1;0.8;0.98;0.59;0.49 (22).
Measurements of apoptosis
The procedure of terminal deoxynucleotide transferase (TdT) assay was taken from Gorczyca et al (23) with slight modification. Briefly, cells were fixed in 1 % paraformaldehyde in PBS followed by 100 % methanol at -20oC. To detect apoptosis the fixed cells were incubated in TdT -solution (5 units in 50 m l) with biotinylated deoxyuridinetriphosphate (Boehringer Mannheim) as substrate for 30 min at 37oC, washed and the cell pellets resuspended in PBS with streptavidin-fluorescein (Amersham), 0.1% Triton X-100 and 3% fat-free milk powder. After 30 min at 4° C, and washing with PBS the cells were treated with 100mg /ml RNAase (Boehringer Mannheim), and stained with propidium iodide (PI) (5m g/ml) for total DNA content. Red fluorescence from the dye propidium iodide (PI) and green fluorescence from labelling DNA-ends in the TdT assay were measured for 104 cells in a Vantage laser flow cytometer (Becton-Dickinson, USA).
Apoptosis in the mouse lymphoma cells was also evaluated in the fluorescence microscope. At least 100 cells were counted in each sample.
Morphological changes in the nuclei were studied in samples stained with propidium iodide as described above. The nuclei were evaluated by counting the percentage of the cells with fragmented nuclei. In the samples stained for the TdT assay, the percentage of the cells with green fluorescence was determined.
Viable and dead cell numbers were determined based on exclusion of trypan blue dye 18 h after light treatment.
RESULTS
Figure 1 indicates that a fraction of the cells treated with bilirubin and light showed signs of apoptosis very rapidly after the end of light treatment. The percentage of apoptotic cells did not increase substantially after 3 h incubation, but an increasing tendency towards disintegration was observed in the microscope after incubation times longer than approx. 18 h. In the later experiments an incubation time of 3 h after the end of irradiation was chosen as standard.
The different methods for observing apoptosis gave corresponding results as shown in table I. It can also be observed that about 60 % of the cells were non-viable cells as indicated by trypan blue staining 18 h after irradiation for 1.5 h in the presence of BSA (table I, fig. 2) and that the percentage of apoptotic cells is smaller than the fraction that can be scored as necrotic by trypan blue staining. This was also the case when the cells were irradiated in the presence of HSA (table II). By comparing the sensitivity to cell death, either by necrosis (fig. 2) or by apoptosis (tables I and II) with the doses leading to significant photooxidation of bilirubin (figure 3)(11), one may draw the conclusion that the cell killing is taking place at light doses causing observable photooxidation of bilirubin.
The effect of splitting up the light dose in fractions was compared with the effect of continuous irradiation in table II. Due to long time incubation in PBS the toxicity in the controls was relatively high compared to the values seen in table I and figure 2 (values for samples treated for 2 h or less in the presence of BSA). No significant difference between cells treated with continuous and split light doses, respectively, can be observed, although there is a tendency of higher toxicity and more apoptosis after split light exposures.
DISCUSSION
The present study has shown that mouse lymphoma cells of the line L5178Y-R can undergo apoptosis and that the development of the reaction is relatively rapid (fig. 1). Early apoptosis in cells in vitro is also induced by other photosensitised reactions, e.g. by photoactivated protoporphyrin (24). However, the development of apoptosis is dependent on the sub-cellular localisation of the photosensitising drugs (25).
Three different methods have been used to measure the fluorescence from the cells, and two different biological characteristics, namely the presence of free DNA-ends and morphological changes of the nuclei, give rise to the differences in fluorescence observed. In a previous paper (11) an indication of double strand breaks in the DNA of mouse 308 epitheloid cells was found, but no gross morphological changes were observed. The choice of LY-R cells in the continuation of our studies was based on the fact that they are sensitive to ultraviolet radiation and hydrogen peroxide, and it has been shown by the authors (11) and by Rosenstein et al (7) that this species and other peroxides may be important for the phototoxic effects of bilirubin. It was shown previously that the phototoxicity is prominent after light doses that cause photooxidation of bilirubin, which is supported by the data in figures 2 and 3. Furthermore, it was shown that a fraction of the phototoxic products leading to cellular damage was formed in the medium outside the cells, and that addition of catalase, but not superoxide dismutase, to the medium reduced the cytotoxicity (8,9). Therefore, it is reasonable to assume that the fact that the LY-R cells undergo apoptosis is a result of hydrogen peroxide production in the cells and in the medium.
The toxicity of bilirubin in the dark is not fully elucidated, but several mechanisms are possible (26). It is probable that the dark toxicity and the phototoxicity of bilirubin act by different mechanisms and that some of the phototoxic reactions are a «type III» -photosensitising effect arising as a result of the toxicity of the photoproducts of bilirubin acting together with the production of H2O2 (7). In cells treated with photoactivated bilirubin, it is to be expected that the cellular damage is a mixture of responses to bilirubin and its photoproducts. The present paper indicate that apoptosis as well as necrosis are possible modes of cell death.
Our data show that the presence of free bilirubin (not bound to albumin) increases the efficiency of the photosensitisation. In figure 2 and the tables I and II it can bee seen that the cells are killed by a lower light dose when suspended in 50 mM BSA instead of a solution of 200 mM HSA. Both solutions contained 160 mM bilirubin, however, the latter containing HSA had a higher molar concentration and most of the bilirubin was bound to the HSA.
The split dose experiments show that there is a possibility that delivery of light at a high dose rate in fractions may give slightly more cellular damage than continuous light exposure, but these experiments are technically difficult to perform and the variance between individual experiments is large. Furthermore, the toxicity in the controls is relatively high. The temperature of the cell suspensions were measured during a typical experiment and was found to be below 37oC. In the future more detailed studies will be performed to see if the tendency observed is significant, and to attempt to explain a possible split dose effect or a dose rate effect. The dose rate during phototherapy may influence the formation of bilirubin isomers. Probably there exists an optimal dose rate where the therapeutic effect is maximum (3) and the detrimental effects of light exposure are of limited significance for the patient.
ACKNOWLEDGEMENTS
This study was supported by The Research Council of Norway.
The high quality technical assistance of Mr. Fredrik Liland is highly appreciated.
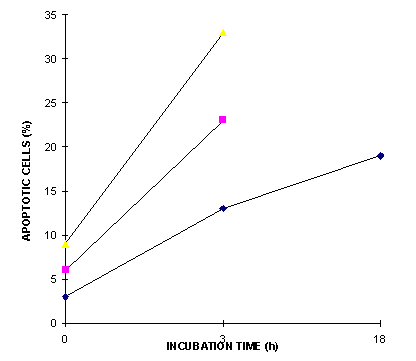
Figure 1: The induction of apoptosis assayed by the formation of fragmented nuclei in mouse lymphoma cells stained with propidium iodide at 0, 3 and 18 hours after blue light irradiation for 1.5 h of 5.5 mW/cm2 irradiance in a solution of bilirubin containing BSA. Results from three independent experiments are shown.
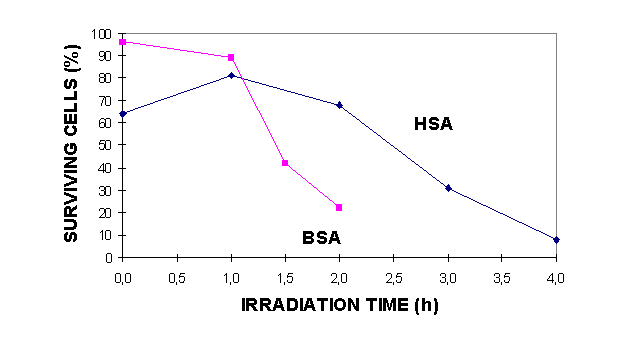
Figure 2: Induction of necrosis in mouse lymphoma cells irradiated with blue light (5.5 mW/cm2) for 0-4 h in solutions of bilirubin and BSA or HSA. Exclusion of trypan blue was assayed after 18 h incubation.
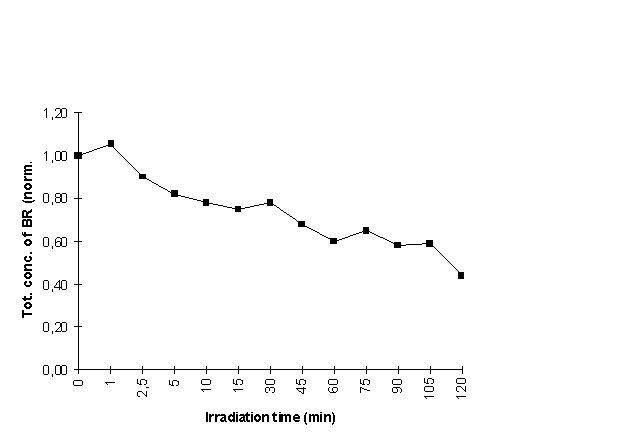
Figure 3: The sum of conc. of ZZ-BR and photoisomers E-lum, Z-lum and ZE/EZ-BR after
irradation times of 0-120 min. Total conc. is normalised to 1.0 prior to irradiation.
| Treatment | Method | Apoptosis (%) | Standard deviation |
| Control | PI | 2.4 | 1.3 |
| Control | TdT, microscopy | 6.7 | *: two experim. |
| Control | TdT, flow cytom. | 4.2 | 3.8 |
| 1.5 h light, BR | PI | 20 | 8.0 |
| 1.5 h light, BR | TdT, microscopy | 40 | 23 |
| 1.5 h light, BR | TdT, flow cytom. | 35 | 25 |
Table I: Apoptosis in mouse lymphoma cells assayed with three different methods. The cells were suspended in a bilirubin solution containing BSA and irradiated with blue light (5.5 mW/cm2). The cells were fixated 3 h after the end of irradiation.
| Sample | Necrosis, trypan blue ( %) | S.E. | Apoptosis, PI (%) | S.E. | Apoptosis, TdT (%) | S.E. |
| I, control, split | 34 | 12.4 | 42.3 | 3.8 | 39.4 | 6.1 |
| I/2, control, continuous | 26 | 9.3 | 35.4 | 5.1 | 26.4 | 5.3 |
| I BR, split | 71.3 | 10.7 | 67.8 | 6.1 | 69.5 | 5.5 |
| I/2 BR, continuous | 61 | 9.6 | 54.9 | 11.5 | 44.9 | 3.9 |
| Dark control | 20.3 | 5.1 | 25.3 | 6.3 | 24.5 | 4.6 |
| Dark BR | 19 | 2.1 | 25.0 | 8.4 | 25.3 | 5.9 |
Table II: Cell necrosis assayed with trypan blue exclusion and apoptosis assayed with two different methods in mouse lymphoma cells. The cells were suspended in a bilirubin solution containing HSA and irradiated with blue light, either 5.5 mW/cm2 (I) for a total of 3 h in three fractions of 1 h each, interrupted by two 1 h periods in the dark, or continuously for 6 h at irradiance of 2.7 mW/cm2 (I/2). In both cases the total light fluence was the same (5.9 J/cm2). The cells were fixated 3 h after the end of irradiation. Mean of three separate experiments with two parallel samples and corresponding standard error are shown.
References
- T.Christensen, J.B.Reitan, Fototerapi av hyperbilirubinemi hos nyfødte. SIS Rapport 1987:6 (In norwegian)
- C. Vecchi, G.P. Donzelli, M.G. Migliorini, G. Sbrana , R. Pratesi, New light in phototherapy. Lancet 14 (1982) August , 390
- K.L. Tan The pattern of bilirubin response of phototherapy for neonatal hyperbilirubinemia. Pediatr. Res. 16 (1982) 670-674
- D. Jährig , A. Berck, P. Meisel, K. Jährig, Die Kalkulation die Phototherapieeffektes unter Berücksichtigung verschiedener Bestrahlungsvarianten, I. Der Einfluss des Therapiemodus auf den Photoeffect, Kinderärzl. Praxis 53 (1985) 171-175
- T.R.C. Sisson, S.C. Glauser, E.M. Glauser, W. Tasman, T. Kuwabara, Retinal changes produced by phototherapy. J Pediatrics 77 (1970) 221-227.
- T. Christensen, J.B. Reitan, G. Kinn, Single strand breaks in the DNA of human cells exposed to visible light from phototherapy lamps in the presence and absence of bilirubin, J. Photochem. Photobiol. B. Biol. 7 (1990) 337-346.
- B. S. Rosenstein, J. M. Ducore, S. W. Cummings, The mechanism of bilirubin-photosensitized DNA strand breakage in human cells exposed to phototherapy light, Mutation Res. 112 (1983) 397-406
- T. Christensen, Cytotoxicity of bilirubin photoproducts, Photobiochem. Photobiophys. 10 (1986) 253-260
- T. Christensen, A. Støttum, G. Brunborg, J.B. Reitan, Unwanted side effects and optimization of phototherapy. In R. H. Douglas, J. Moan and F. Dall'Acqua (eds.), Light in biology and medicine. Volume 1, Plenum Press, New York and London, 1988, pp. 153-159
- T. Christensen, G. Kinn, T. Granli, I. Amundsen, Bilirubin and light effects on cells. 12 th International congress on photobiology, Wien 1. - 6. Sept. (1996) (abstract nr. O 46)
- T. Christensen , G. Kinn, I. Gradzka, A. Jaworska, E. B. Roll, Photosensitising effects of bilirubin First Internet Conference on Photochemistry and Photobiology, November 17. - December 12. 1997, Internet Journal of Science Biological Chemistry, vol. 3 (1997), http://www.photobiology.com/v1/contrib.htm
- R. Stocker, B.N. Ames, Potential role of conjugated bilirubin and copper in the metabolism of lipid peroxides in bile, Proc. Natl. Acad. Sci. USA 84 (1987) 8130-8134.
- R. Stocker, A.N. Glazer, B.N. Ames, Antioxidant activity of albumin-bound bilirubin, Proc. Natl. Acad. Sci. USA 84 (1987) 5918-5922.
- R. Stocker, Y. Yamamoto, A.F. McDonagh, A.N. Glazer, B.N. Ames, Bilirubin is an antioxidant of possible physiological importance, Science 235 (1987) 1043-1046
- J.Z. Beer, E. Budzicka , E. Niepokojczycka et al., Loss of tumorigenicity with simultaneous changes in radiosensitivity and photosensitivity during in vitro growth of L5178Y murine lymphoma cells. Cancer Res. 43 (1983) 4736-4742
- D.E. Godar, A.D. Lucas , Spectral dependence of UV-induced immediate and delayed apoptosis: The role of membrane and DNA damage. Photochem. Photobiol. 62 (1995) 108-113.
- D. Separovic, J. He , N.L. Oleinick, Ceramide generation in response to photodynamic treatment of L5178Y mouse lymphoma cells. Cancer Res (1997) 1717-1721.
- H. Tauchi , S. Sawada, Analysis of mitotic cell death by radiation in mouse leukaemia L5178Y cells: Apoptosis is the ultimate form of cell death following mitotic failure. In. J. Radiat. Biol. 65 (1994) 449-455.
- E. Bouzyk , T. Iwanenko, N. Jarocewicz, M. Kruszewski, B. Sochanowicz, I. Szumiel, Antioxidant defense system in differentially hydrogen peroxide sensitive L5178Y sublines. Free Radical Biology & Medicine 22 (1997) 697-704.
- J.Z. Beer, K.M. Olvey, S.A. Miller, D.P. Thomas, D.E. Godar, Non-nuclear damage and cell lysis are induced by UVA, but not UVB or UVC, radiation in three strains of L5178Y cells. Photochem. Photobiol. 58 (1993) 676-681.
- T. Christensen, G. Kinn, T. Granli, I. Amundsen, Cells, bilirubin and light. Formation of photoproducts and cellular effects at defined wavelengths. Acta Pædiatr. 83 (1994) 7-12
- G. Agati, F. Fusi, S. Pratesi, P. Galvan, G.P Donzelli, Bilirubin photoisomerization products in serum and urine from a Crigler-Najjar type I patient treated by phototherapy, J. Photochem. Photobiol. 47 (1998) 181-189
- W. Gorczyca, H. Gong, Z. Darzynkiewicz, Detection of DNA strand breaks in individual apoptotic cells by the in situ terminal deoxynucleotidyl transferase and nick translation assays, Cancer Res. 53 (1993) 1945-1951
- B.B. Noodt, K. Berg, T. Stokke, Q. Peng, J.M. Nesland, Apoptosis and necrosis induced with light and 5-aminolaevulinic acid-derived protoporphyrin IX, Br.J. Cancer 74 (1996) 22-29
- D. Kessel, Y. Luo, Y. Deng, C.K. Chang, The role of subcellular localization in initiation of apoptosis by photodynamic therapy. Photochem Photobiol 65 (1997) 422-426
- J.W. McDonald, S.M. Shapiro, F.S. Silverstein, M.V. Johnston, Role of glutamate receptor-mediated excitotoxicity in bilirubin-induced brain injury in the Gunn rat model, Exp. Neurol. 150 (1998) 21-29