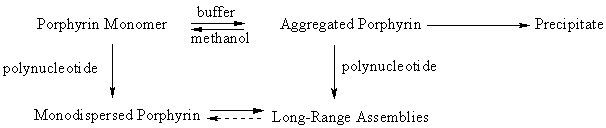| Papers and Posters | Site Home Page |

Porphyrin assemblies on poly(dG-dC)2 and poly(dA-dT)2
![]()
K. Lang, D. M. Wagnerová,
P. Anzenbacher Jr., V. Král P. Kapusta P. Kubát |
![]()
Abstract The present paper describes synthesis and spectroscopic characterization of novel cationic meso-tetraphenylporphyrins bearing two (trans) (P2) or three (P3) triphenylphosphonium substituents. P2 and P3 undergo aggregation in aqueous solutions. Newly formed components with absorption bands around 406 nm and 430 nm were attributed to distinct porphyrin aggregates. In the presence of poly(dA-dT)2 and poly(dG-dC)2 the porphyrins are bound and self-organized onto long-range assemblies on the strand exterior as follows from absorption, fluorescence, CD, transient and resonance light-scattering (RLS) spectroscopies. Induced circular dichroism and intensive RLS indicate exciton coupling occurring after binding to the chiral environment on the nucleic acids exterior. The similarity of CD spectra of P2 on poly(dG-dC)2 and poly(dA-dT)2 suggests that the binding geometry is essentially independent of nucleic acid sequences. The fluorescence lifetime of about 4 ns was attributed to the long-range assemblies. The distinct CD spectra of P3 bound on GC or on AT base-pair regions reveal that the number of the porphyrin substituents determines how closely the porphyrin can approach to the nucleic acid helix. The higher hydrophobicity of P2 is manifested by higher aggregation tendency in buffer and consequently by more extensive self-organization on the polynucleotide exterior. Keywords: Porphyrin, Polynucleotide, Aggregation, Long-range assembly, Excited states
|
![]()
|
Porphyrins have potential biological applications due to their photosensitizing ability and selectivity for DNA cleavage upon appropriate conditions. In general, water-soluble cationic porphyrins bind to synthetic and natural nucleic acids. The complexation has been studied by biochemical techniques and static and time-resolved spectroscopy methods including absorption, fluorescence, circular dichroism, resonance light-scattering, Raman, EPR and NMR [1-14]. Most studies have focused on meso-tetrakis(4-N-methylpyridyl)porphyrin (TMPyP) [1-8] and corresponding metalated derivatives [2,4,9,13,14]. It is commonly believed that TMPyP discriminates between GC-rich and AT-rich regions [6,7,9]. Three main binding modes, controlled by the DNA base sequence, porphyrin shape and charge, have been recognized so far [5-7,9]. Fiel et al. [1] were first to suggest that some porphyrins, which can exist at least temporarily in a planar conformation, intercalate into DNA base pairs. Intercalation of TMPyP occurs in GC-rich regions with binding constants of about 106 M-1 [1,2,4,5]. External (groove)binding is typical for sterically constrained porphyrins having axial ligands on the central ion of metalloporphyrins or bulky substituents on the porphyrin moiety [2,3,15]. The apparent binding constants are of the same order of magnitude as those for intercalation [4,10,15]. The third mode, external binding with self-stacking was originally proposed for meso-tetra(p-N-trimethylanilinium)porphyrin at large porphyrin/DNA ratios to explain two CD Cotton effects of opposite signs in the Soret region of the porphyrin [9,10]. The formation of organized long-range assemblies on the exterior of the helical polymer was also observed for meso-bis(4-N-methylpyridyl)diphenylporphyrins [11,12,16]. In spite of extensive studies, the binding affinity to nucleic acids has been examined for limited number of cationic porphyrins. In this respect, purposeful functionalization of the porphyrin moiety offers a number of porphyrins differing in charge, substituents, hydrophobicity, etc. Lipophilic substituents may make it easier for the porphyrin to pass through or accumulate in biomembranes, may change its affinity to nucleic acids and so influence the site recognition. Some of us have developed a general strategy for the synthesis of positively charged porphyrins with easily controlled hydrophobicity [15] and used it for the synthesis of novel triphenylphosphonium porphyrins. Aggregation [15,17], physicochemical properties and interaction with CT DNA [15] of 5,10,15,20-tetrakis(a-triphenylphosphonium-p-tolyl)porphyrin have already been reported. The conditions for long-range assembling on the nucleic acid exterior [11,12] and its kinetics [16] have been studied for meso-bis(4-N-methylpyridyl)diphenylporphyrins. Such supramolecular systems are of great interest because the enforced helical structures of photoactive and redoxactive molecules can be utilized for construction of molecular-based devices. They can also serve as sensitive supramolecular sensors of DNA sequences. Here we wish to report the synthesis of novel triphenylphosphonium porphyrins depicted in Figure 1. Two (trans) (P2) and three (P3) positive charges in conjunction with lipophilic phenyl substituents cause aggregation properties strongly sensitive to ionic strength; hence, the porphyrins can be expected to suit well for self-assembling on nucleic acid exterior. The long-range assemblies of the title porphyrins on poly(dG-dC)2 and poly(dA-dT)2 were characterized in detail by static and time-resolved spectroscopic techniques. |
|
Material. Double-stranded poly(dG-dC)2 and poly(dA-dT)2 (Pharmacia Biotech) were dissolved in 20 mM phosphate buffer, pH 7.0, containing 100 mM NaCl and stored at -18 oC. The concentrations of poly(dG-dC)2 and poly(dA-dT)2, calculated in base pairs, were determined spectrophotometrically using molar absorptivity e254 = 1.68 × 104 M-1 cm-1 [18] and e262 = 1.32 × 104 M-1 cm-1 [19]. All experiments were performed in 20 mM phosphate buffer (pH 7.0, 100 mM NaCl) at different porphyrin/base pair molar concentration ratios R at room temperature. Meso-tetrakis(4-N-methylpyridyl)porphyrin tetratosylate (TMPyP) and meso-tetrakis(4-sulfonatophenyl)porphyrin tetrasodium salt (TPPS) were purchased from Porphyrin Products, Utah, USA. The stock solutions of P2 and P3 (Fig. 1) were prepared in methanol (HPLC grade, Aldrich) and diluted with buffer prior to use giving concentrations below 5 mM. General procedure for preparation of triphenylphosphonium porphyrins. A microvial with a septum sealed inlet was charged with 0.26 g of triphenylphosphine (1.0 mmol) and corresponding (a-bromo-p-tolyl)porphyrin (0.033 mmol), evacuated, filled with argon and heated to 110 °C. The melt was maintained at 110 °C for 12 hr upon stirring. Absolute toluene (5.0 ml) was added through the septum and the resulting suspension was cooled to 70 °C. Dark brown-violet precipitate was filtered off, washed by warm toluene and dried in vacuum. The porphyrins with two (trans) (P2), three (P3) and four a-triphenylphosphonium-p-tolyl substituents were characterized by elemental analysis, UV/VIS, NMR and FAB mass spectrometry. On the basis of preliminary experiments P2 and P3 were selected for further studies since they exhibit appropriate aggregation properties in aqueous solutions. 5,15-Bis(a-triphenylphosphonium-p-tolyl)-10,20-bis(p-tolyl)porphyrin
dibromide salt (P2): 5,10,15-Tris(a-triphenylphosphonium-p-tolyl)-20-(p-tolyl)porphyrin
tribromide salt (P3): Methods. UV-VIS absorption spectra were measured on Perkin Elmer Lambda 10 and Philips PU 8720 spectrophotometers. Circular dichroism spectra were measured on a Jobin Yvon-Spex CD 6. All spectra were obtained by averaging 3 accumulations recorded with steps of 0.5 nm (1 s integration time). Steady state fluorescence emission spectra were recorded on a Perkin Elmer LS 50B luminescence spectrometer. The samples were excited at the visible Qy(1,0) band (520 nm), which is less influenced by binding than the Soret band. Relative fluorescence quantum yields were obtained by comparison of the total porphyrin fluorescence intensity in the presence of polynucleotide and in methanol using the correction factor n2(water)/n2(methanol) (n is the index of refraction). Resonance light-scattering experiments (RLS) were conducted using simultaneous scans of the excitation and emission monochromators through the range of 300 - 600 nm. Fluorescence decay kinetics were measured on an Edinburgh Instruments FS/FL900 steady-state/time-resolved spectrofluorimeter, using hydrogen filled nF900 pulsed nanosecond flash lamp excitation and time-correlated single photon counting detection. The thermostatted samples (20 oC) in 1 cm quartz cuvettes were excited at 420 nm (20 nm bandwidth), the fluorescence was collected at 652 nm (10 nm bandwidth). The instrument response function (half width 1.2 ns) was measured using a ludox scattering solution. Fluorescence lifetimes were determined by non-linear least-squares iterative re-convolution fitting of the raw data. The quality of the fit was evaluated by inspection of the residual distribution and the reduced c2 value. Laser flash photolysis experiments were performed with a Lambda Physik FL 3002 dye laser (413 nm, output ~ 2 mJ/pulse, pulse width ~28 ns). Transient spectra were recorded within 300 - 600 nm on a laser kinetic spectrometer (Applied Photophysics). The time profiles were usually probed at 470 nm (TMPyP, P3), 440 nm (TPPS) for the triplet states and at 420 nm, 430 nm, 444 nm (TMPyP), 414 nm (TPPS) and 430 nm (P3) for the recovery of the bleached ground state using a 250 W Xe lamp equipped with a pulse unit and a R928 photomultiplier. The samples were saturated by air or by oxygen, in some cases oxygen was removed by purging the solution with argon. |
|
3.1. Aggregation in aqueous solution The porphyrins P2 and P3 were spectroscopically investigated up to 90 m M in methanol (Table 1, Table 2). The absorption in the Soret region at 416 nm and in the visible region at 520 nm - 650 nm obey the Beer's law, thus indicating monomeric forms. At variance, the Soret band in distilled water with ionic strength I ~ 0 manifests a strong hypochromism (~ 50% for P3) concomitant with a large broadening (Fig. 2a). A bathochromic shift is observed in the visible region (Table 2). The broad Soret band can be resolved by the second derivative into three distinctive components peaking at 406 nm, 418 nm and 430 nm (variations ± 2 nm) (Fig. 2d). Notable is the disappearance of the 406 nm component when methanol is added (about 30 % for P2, 10 % for P3). Adjustment of ionic strength by NaCl (100 mM) exerts a strong influence on the Soret band. In the case of P2, the second derivative shows complete substitution of the 418 nm component by the intensive band at 424 nm converting to that at 436 nm in aged solutions. In contrast, the absorption spectra of less hydrophobic P3 show the 418 nm component even though its intensity is considerably lowered when compared to solutions of I ~ 0. On the other hand, the intensity of the 430 nm component is eminent. These changes are accompanied by a decrease of fluorescence intensities, whereas no shifts of the emission wavelengths can be observed (Table 1). Fluorescence excitation spectra confirm marked presence of components around 406 nm and 430 nm having much lower fluorescence quantum yields than the monomer. It should be emphasized that fluorescence intensities decrease with aging and with vigorous stirring of the solution. In some cases, precipitate can be observed in aged solutions. Significant influence of ionic strength and methanol on the Soret band is most likely due to formation of porphyrin dimers and higher aggregates. A strong exciton coupling between neighboring monomeric dye units can, according to Kasha et al. [20], lead to excited state energy level splitting that manifests itself in a splitting of the Soret band. The blue-shifted absorption band located at 406 nm is assumed to belong to an aggregate (dimer) in which the porphyrins are stacked face-to-face. This band is considerably broadened due to a range of different conformations [21]. Such aggregates can easily be monomerized in mixed solutions with organic solvents [22]; this process was also observed for the porphyrins upon addition of methanol. The red-shifted component is associated with an aggregate, a J-aggregate, in which the centers of porphyrin moieties are displaced from each other [20,21,23] with transition dipoles inclined to interconnected axis by an angle smaller than 54.7o [20]. The geometry of J-aggregates is controlled by ionic strength and by steric hindrance imposed by pendant substituents. By inference, the changes of electrostatic interactions between porphyrin moieties during the growth of J-aggregate could slightly shift the absorption band as observed for P2 (see above). The formation of the extended assemblies was also confirmed by the appearance of RLS spectra. A scattering species has lmax» 449 nm (Fig. 3a) and RLS intensity grows with increasing absorption at about 436 nm for P2 and at 430 nm for P3. The third resolved absorption band at about 418 nm belongs evidently to the monomer since it is similar to methanol solutions. We applied controlled ionic strength (100 mM NaCl) in order to maintain polynucleotides double-stranded during binding experiments and to eliminate the effects of ionic strength variations on noncovalent interactions of the porphyrins (self-organization, binding to polynucleotides). 3.2. Spectral properties of porphyrin-polynucleotide complexes When the porphyrin monomers of P2 and P3 are added into the solutions of poly(dA-dT)2 or poly(dG-dC)2, broadening of the Soret bands and bathochromic shifts of the visible bands are similar to spectral changes occurring in buffer (Table 1, Table 2, Fig. 2). The fluorescence emission spectra consist of two bands with maxima little affected by polynucleotides (Table 1), however, the emission intensity is markedly reduced. Thus, P2/poly(dG-dC)2 (R Ł 0.18) and P3/poly(dG-dC)2 (R Ł 0.22) exhibit the relative fluorescence quantum yields of 0.33 and 0.20, respectively, related to the corresponding monomer in methanol. A notable effect of polynucleotides is in strong contrast with porphyrin behavior in buffer solutions - absorption spectra and fluorescence quantum yields are independent on R up to about 0.2 and stable for hours after the solution had been prepared. In addition, no interfering adsorption of the porphyrins on cell walls has been observed. It clearly indicates nearly quantitative binding to the polymers up to R » 0.2 due to availability and a large number of binding sites, and appropriate porphyrin hydrophobicity. The second derivative of the Soret band resolves three components (Fig. 2e). The peak of the bound monomer at about 420 nm is bathochromically shifted by a few nm compared to buffer solutions (Table 1). When the P2 and P3 spectra are compared, the former shows only a shoulder of the monomer given by its higher hydrophobicity while the latter shows well-resolved monomer bands at 418 nm and 422 nm, respectively. Similar small shifts of the monomer bands were reported for externally bound tetracationic porphyrins onto natural or synthetic DNA, while intercalation of porphyrin led to more substantial spectral changes [2-4,9]. The blue-shifted band at 406 nm is not affected by polynucleotide, however, the red-shifted absorption around 437 nm unprecedentedly increases upon binding (Fig. 2). The observed spectral variations indicate that the porphyrins are most likely self-assembled on the polymer matrix and that the monodispersed porphyrin is a minor component. Here the term monodispersed denotes the bound monomer. The blue-shifted bands suggest the disposition of the aggregated porphyrins to be face-to-face even upon binding to polynucleotide. This tendency is more explicit for P2 of R Ł 0.1 (Fig. 2b,c). As stated by Pasternack et al. [11,12], the red-shifted absorption is due to porphyrin-porphyrin interactions within long-range assemblies on the polymer exterior. Their formation can be mediated by Coulombic attraction between backbone phosphates and positively charged porphyrin substituents at appropriate ionic strength and R, the conditions at which porphyrins have high tendency to aggregate in buffer [11,12,24]. Extended porphyrin-porphyrin assemblies can be visualized in RLS spectra [11,12,24-26]. In accord with it, we observed the appearance of RLS bands in the Soret region with maxima of the scattering particles at 437 nm. The higher disposition of P2 than P3 to aggregate is manifested by more intensive RLS under the same conditions (Fig. 3b,c). It is worth recalling that the amount of scattered light is directly proportional to the volume of aggregate [12,25]. Hence, the high RLS intensity (Fig. 3b,c) compared to buffer solutions (Fig. 3a) expresses the induced compaction of porphyrins into extended assemblies on the polynucleotide matrix. In buffer the porphyrins are achiral with no circular dichroism spectra (CD). Binding to polynucleotides induces appearance of CD spectra (Table 1). This is indicative of helical alignment of the porphyrin transition dipole moments due to interaction of porphyrins with chiral environment [1-3,13,14,27,28] or due to exciton coupling within chiral assemblies [10-12,29]. An induced optical activity is a characteristic property for distinct binding modes: a single negative peak for the whole Soret band characterizes monodispersed intercalated porphyrin and a single positive Cotton effect belongs to monodispersed externally bound porphyrins at DNA sites. Bisignate patterns of induced signals document exciton coupling of closely spaced porphyrin units. P2/polynucleotide shows a split Cotton effect in the Soret region and low molar De with positive peak at about 422 nm and negative peak at about 438 nm, though the lmax of the Soret band attributed to aggregates is at 437 nm, near mid-point of the two CD peaks at 432 nm (Fig. 4a, Fig. 5a). When resolved into three Gaussian curves in wavenumber units, two bands show equal rotational strength of opposite signs which is the characteristic for exciton split Cotton effects [30]. The third positive Gaussian curve at 413 nm is indicative of the monodispersed porphyrin on the polynucleotide exterior. At variance, induced CD spectra of P3 are affected by the polynucleotide composition. To achieve a unambiguous resolution for the P3/poly(dA-dT)2 spectrum with two positive peaks at 415 nm, 437 nm and a negative peak at 427 nm (Fig. 4b) is difficult. Because the absorption spectra confirm higher contribution of the monodispersed porphyrin that for P2, the resulting CD spectra are probably given by superposition of external binding to the major and minor polynucleotide grooves and by self-assembly. On the other hand, only a single bisignate signal is observed for P3/poly(dG-dC)2 (Fig. 5b). Optical activity was investigated up to R » 0.07 and no significant changes of the peak positions and De were found. 3.3. Time-Resolved Fluorescence The fluorescence decay of P2 and P3 in methanol is clearly monoexponential with lifetimes of 9.7 ns and 9.4 ns (Table 3) indicating the exclusive presence of the porphyrin monomer. On the contrary, analysis of the decay profile of P3 in 20 mM phosphate buffer (pH 7.1, without NaCl in order to slow down aggregation) shows that it can best be fitted to a biexponential function with a fast component tf1 = 1.8 ns and a second component tf2 = 11.8 ns. We were unable to measure P2 in buffer due to its fast aggregation even in the absence of NaCl. In the presence of polynucleotides, the fluorescence intensities of P2 and P3 decrease biexponentially as well, with lifetimes of approximately 4 ns, 15 ns and 2 ns, 12 ns, respectively (Table 3). In no case was the quality of the fit significantly improved by using triexponential fitting function, thus validating the use of a biexponential function. Possible explanation of our results is a partition of the porphyrins between two
distinct populations differing in fluorescence lifetimes. The lifetimes of P3/poly(dA-dT)2
and P3/poly(dG-dC)2 are almost coincident with the lifetimes of P3 in
buffer (Table 3). However, the amplitude of the short-lived
component doubles in the presence of polynucleotides, i.e. under conditions where
long-range assemblies are predominant (see above for absorption, RLS and CD results).
Referring to the considerable shortening of lifetimes upon porphyrin aggregation in
solutions [31,32] we conclude that the short-lived components
belong to the porphyrin assemblies and the long-lived components to corresponding monomers
(see 3.4.). The lifetimes of the assemblies of P3 (~ 2 ns) are reduced
approximately by a factor of 2 to those of P2, regardless of G-C or A-T sequences.
It confirms that the lifetimes are given by inherent properties of the title porphyrins,
which are not affected by binding to the polymer exterior. Moreover, other possible
photoinduced processes like electron transfer between porphyrin and the guanine residues [8,33] are excluded. The relative contributions of components do
not change with R within experimental error as follows from Table
3. This is in agreement with our finding that the fluorescence quantum yields are not
affected for R up to 0.2. It also means that the size distribution of the
long-range assemblies for 3.4. Triplet State Formation and Effect of Complexation The behavior of the porphyrin triplet states is affected by binding to polynucleotide (Table 4). The triplet states of intercalated and externally bound TMPyP to poly(dG-dC)2 and poly(dA-dT)2 exhibit longer lifetimes in the absence [8] and in the presence of oxygen. On the other hand, anionic TPPS does not interact with polynucleotides since absorption, fluorescence spectra and relaxation processes following the excitation are not changed in their presence (not shown). Evidently, electrostatic repulsion between TPPS and the backbone phosphates does not allow any binding. We found that the triplet states of P2 and P3, due to self-assembling, were produced with remarkably low quantum yields. Therefore, it is difficult to analyze their behavior. The triplet lifetimes of P3 on poly(dG-dC)2 are nearly oxygen independent (Table 4) though in general, the triplet states of porphyrins are quenched by oxygen efficiently. Evidently, our results point to a very low amount of the monodispersed porphyrin on the poly(dG-dC)2 exterior. 3.5. Helical Assemblies of the porphyrins on the polynucleotide exterior Porphyrins are generally prone to aggregate in solution [34]. In most cases aggregates appear to be a mixture of clusters without uniqueness in the structure and number of monomer units. The porphyrin planes are stacked in a slipped cofacial arrangement with interplanar distances ranging from 0.35 to 0.40 nm. The geometry of aggregates is controlled mainly by Coulombic repulsion and by the van der Waals contact between the porphyrin rings [35]. Exciton coupling between monomer units is manifested by changes in absorption, fluorescence, CD and RLS spectra thus giving information on the aggregate geometry. Spectral and photophysical properties of P2 and P3 in buffer are typical for aggregates. The results presented here show that the high porphyrin concentration and ionic strength are not essential for aggregation. The process occurs at concentrations as low as 1 m M and at I ~ 0. It expresses the porphyrin hydrophobicity caused by triphenylphosphonium substituents. The complex spectral changes with no isosbestic points in absorption spectra suggest two types of cofacial aggregates and slow aggregation with continuous increase of the aggregate size. Monomerization occurs after addition of methanol. The multiple processes can be schematically represented as follows
The absorption and fluorescence spectra of P2 and P3 in buffer and in the presence of polynucleotides are similar and do not reveal binding phenomena. Apparently, the polymer matrix has little effect on the spectroscopic properties of the self-organized porphyrins and on fluorescence lifetimes. In contradiction to it, bound P2 and P3 provide high intensity RLS profiles in the Soret region. Because the intensity of RLS signals is proportional to the volume of scattered particles it indicates long-range assemblies organized on the nucleic acid exterior. While in buffer much less intensive RLS is not connected with any optical activity of the porphyrins, binding to polynucleotide induces appearance of circular dichroism - the conservative CD spectra point to exciton coupling occurring among chromophores in a chiral environment. These findings indicate self-organization of the porphyrin units onto long-range assemblies on the strand exterior. Thus, the alignment of the porphyrin transition dipole moments can match the helical arrangement of the phosphate anions on the polynucleotide backbone due to electrostatic attraction between phosphates and cationic substituents. The presence of bulky pendant substituents on P2 and P3 imposes steric limitations and excludes intercalation at GC sites, the dominant binding mode for TMPyP and its planar metal complexes. Summing up, the porphyrins undergo outside binding along the nucleic acid surface with dominant formation of long-range assemblies. Furthermore, we have also evidence of a minor component - the monodispersed porphyrin. The binding phenomena are sketchily presented in the generalized scheme above. When comparing P2 and P3 at the same concentration level, the higher hydrophobicity of P2 is manifested by higher tendency of aggregation in buffer and consequently by more extensive self-organization on the polynucleotide exterior. The complexes P2/poly(dG-dC)2 and P2/poly(dA-dT)2 exhibit similar CD spectra independent of the nucleic acid sequence. The biexponential fluorescence decay under conditions, where binding is practically quantitative, arises from at least two types of porphyrin forms. The relative contribution of the major component with lifetime of about 4 ns, ascribed to the long-range assembly, shows that self-organization occurs dominantly even at low R values at which the porphyrin could be better dispersed. It suggests that P2 is nonspecifically bound in the essentially same chiral environment without discriminating between the nucleic acid sites. When R are increased, the assembly grows while contribution of the monodispersed porphyrin remains nearly constant. Absorption, fluorescence and RLS results exhibit little changes for P3 when bound to poly(dG-dC)2 and poly(dA-dT)2. The differences in CD spectra, however, reveal different electronic environment. It shows the importance of the total substituent volume since it determines how closely the porphyrin can approach the nucleic acid helix and the extent of exciton coupling within the assembly. Since GC base-pair regions are more rigid relative to AT base-pair regions [36] the deformations required to stabilize bound molecules are more difficult to be achieved in poly(dG-dC)2. That might be the reason why P3/poly(dG-dC)2 gives only simple conservative CD spectrum similar to P2/poly(dG-dC)2 and P2/poly(dA-dT)2. The formation of noncovalently attached porphyrin arrays on the polynucleotide exterior indicates self-organization of these chromophores into chiral domains. The coverage of the exterior can evidently be tuned by the size variation of interacting cationic substituents. This work was supported by the Grant Agency of the Czech Republic (No. 203/96/1322, 203/99/1163). We thank Dr. H. Votavová for measuring CD spectra. 1. R. J. Fiel, J. C. Howard, E. H. Mark, N. Datta Gupta, Nucleic Acids Res. 6
(1979) 3093. |