| Papers and Posters | Site Home Page |
The solid-state E/Z-photoisomerization of 1,2-dibenzoylethene
Gerd Kaupp*, Jens Schmeyers
Fachbereich 9 - Org. Chemistry I - University of Oldenburg, Germany
http://kaupp.chemie.uni-oldenburg.de
Introduction
E/Z-photoisomerizations of polyenes in confined media are of importance in biology
(e.g. of rhodopsin or of the yellow protein). E/Z-isomerizations of C=C double
bonds in the highly confined crystalline state have been summarized and their solid-state
mechanisms investigated with AFM.[1,2] Long-range
molecular movements were observed that correlate with the crystal structure. Chemically,
rotational mechanisms were judged possible for Z-2-benzylidene-butyrolactone and Z-1,2-bis-1-naphthyl-ethene
by molecular modelling in fixed lattices with the criterium that no van der Waals short
distances below 75 % of their normal values should be allowed up to a total of 90°
rotation.[1] We now address the case of E-1,2-dibenzoyl-ethene
(trans-DBE; P21/c) which was reported to give the
Z-isomer (cis-DBE; P21) in a single
crystal to single crystal manner[3] even though the
molecular shapes change drastically and also the space group changes. The claim was based
on a Weissenberg photograph; the reverse photoreaction was not occuring; the yields were
not given. Neither the crystal lattice of trans-DBE[3]
nor the one of cis-DBE[4] allow for internal
rotations with only moderate van der Waals interactions and the reaction cannot be
topotactic according to the <4 % lattice change criterion.[5]
We therefore revisited the solid-state photolysis of DBE and report our new findings.
Experimental
Solid-state photolyses were performed using a high pressure mercury lamp (Hanau HPK 125)
with a bandpass filter, Wertheimer UVW-55, isolating the 365 nm emission (2.8 % 334 nm;
<1% transmission at 315 nm and 410 nm) or a Pyrex filter. In the AFM measurements[1] a monochromator was used for isolating the 405 nm emission
(bandpass 9.6 nm). The irradiations on the AFM stage were performed from 20 cm distance or
with a lens focussing the nearly parallel beam to the mounted crystal. Yield
determinations used 1H-NMR-analysis. Crystals of trans-
and chromatographed cis-DBE were grown from ethanol and acetone respectively, by
slow evaporation.
Results
Irradiation of trans-DBE in 1 g quantities for 16 h (Pyrex) or 9 h (Pyrex or
UVW-55) gave a 30 % or 17 % yield of cis-DBE. The crystals became turbid and
disintegrated. Irradiation of the (001)-surface of trans-DBE at 405 nm in the
absorption tail of the slighthly yellow crystals gave the expected surface features due to
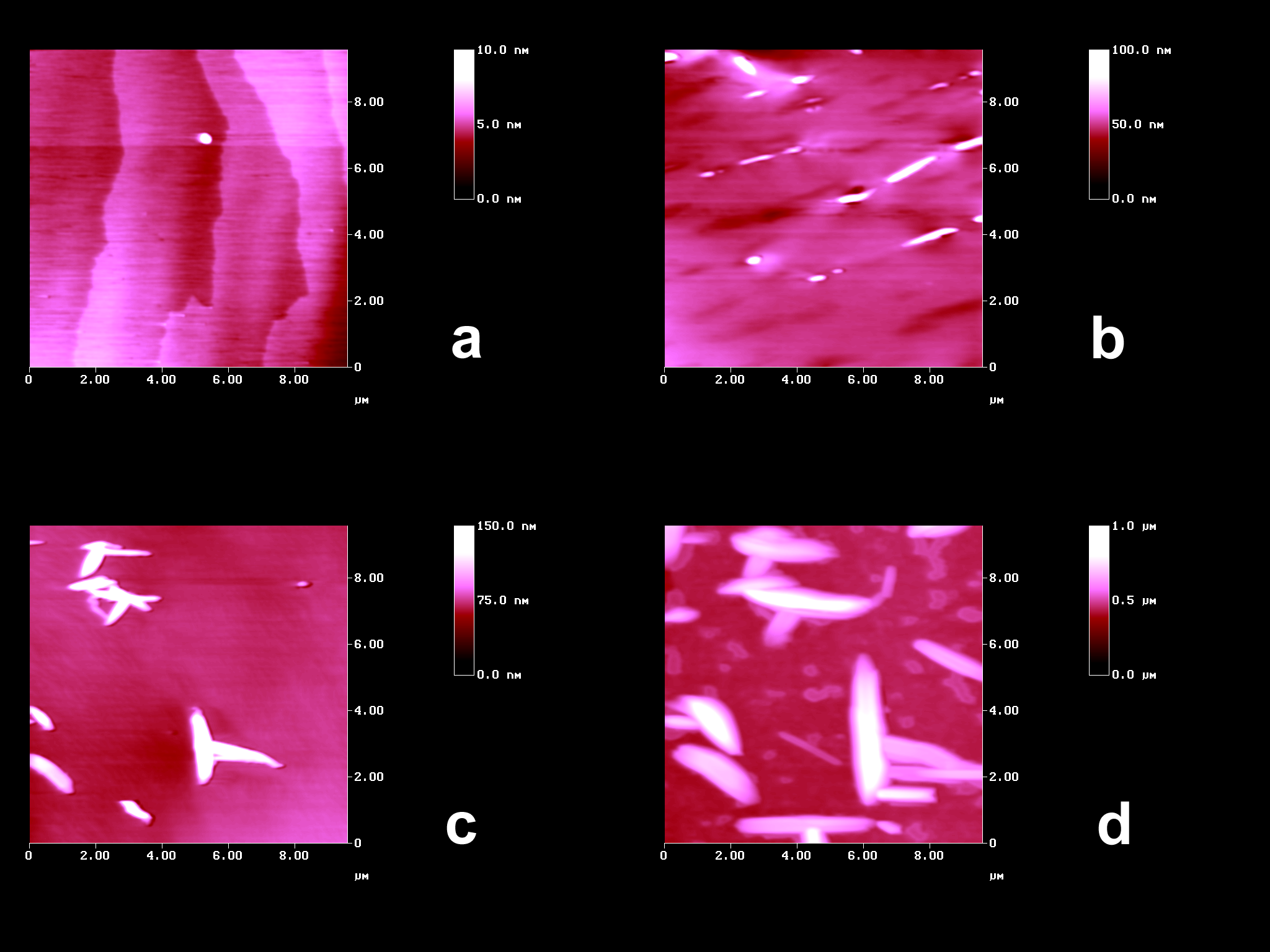
the long-range molecular movements as shown in Figure 1b.
Click on the image for better Quality
A VRML-Plugin (e.g. Cosmo-Player) is required to view the following images !
VRML image of Fig 1 a (low resolution, zipped - 45 kB)
VRML image of Fig 1 b (low resolution, zipped - 43 kB)
VRML image of Fig 1 c (low resolution, zipped - 43 kB)
VRML image of Fig 1 d (low resolution, zipped - 59 kB)
Figure 1. AFM topographies of trans-DBE on (001): (a) fresh with molecular steps; (b) after irradiation at 405 nm; (c) after 30 min irradiation (HPK 125, UVW-55 filter); (d) after 120 min irradiation (HPK 125, UVW-55 filter). The orientations of the crystals are different in (a,b) and (c,d).
The occurence of far-reaching anisotropic molecular movements can be directly seen. Vertical floes align along [010] in Figure 1b. In the more intense shorter wavelength irradiations with a differently oriented crystal (66°), very high floes are formed that continue to grow, however, their orientation is no longer uniform. One direction of preference is still along the b-axis however, further directions are used equally well in Figures 1c,d. Apparently, we see phase rebuilding[1] in Figure 1b and already phase transformation[1] in Figures 1c,d. The latter step is less strictly guided by the initial crystal packing than the former.
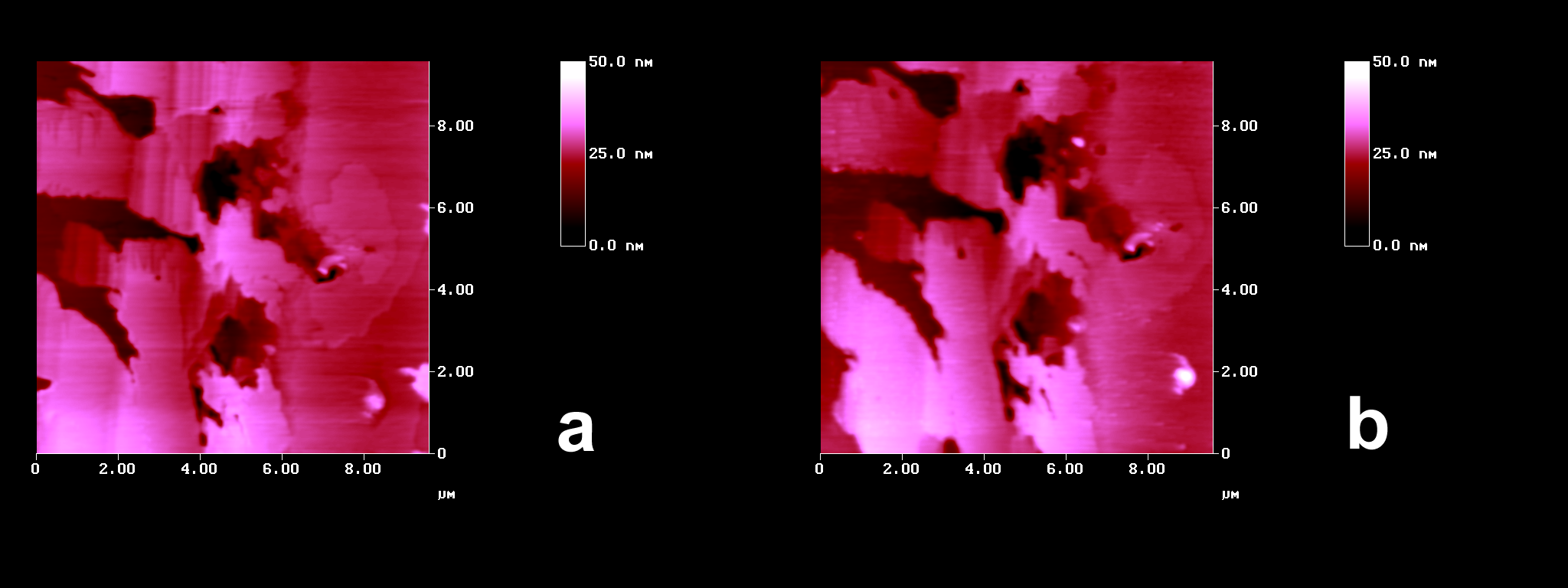
The photostability of crystalline cis-DBE was also probed with the enormous sensitivity of the AFM. The images in Figure 2 show, that there is apart from some slight smoothening no significant change
Click on the image for better Quality
A VRML-Plugin (e.g. Cosmo-Player) is required to view the following images !
VRML image of Fig 2 a (low resolution, zipped - 63 kB)
VRML image of Fig 2 b (low resolution, zipped - 63 kB)
Figure 2. AFM topographies of cis-DBE: (a) fresh, the depressions are 10 to 15 nm down; (b) after 100 min irradiation (HPK 125, Pyrex-filter), showing some smoothening in the 0.5 nm range and the same height of depressions as before.
upon intense Pyrex-filtered irradiation in accordance with the literature report[3] and our preparative tests. This fact is unusual, because that type of solid-state photoisomerizations usually proceeds from the more crowded to the less crowded stereoisomer. We thus address the questions (i) why is trans-DBE photoreactive and (ii) why is cis-DBE not photoreactive in the crystalline state.
Discussion
The interpretation of anisotropic molecular movements and photoreactivities in the
solid-state requires a detailed study of the crystal packing. In sterically demanding
cases like E/Z-isomerizations one has to judge how any free space in the crystal
might be used for the reaction to occur and to start the phase rebuilding. These questions
can be answered by use of the stereoscopic packing diagrams in Figures 3 and 4 that by
necessity deal with rather large sections of the crystals.
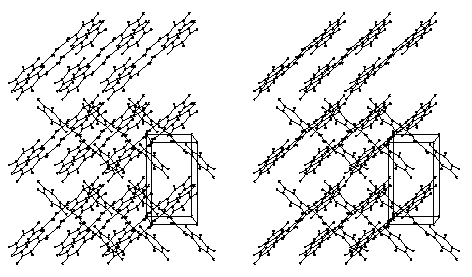
Figure 3. Stereoscopic packing diagram of trans-DBE facing (001); the a-axis is vertical.
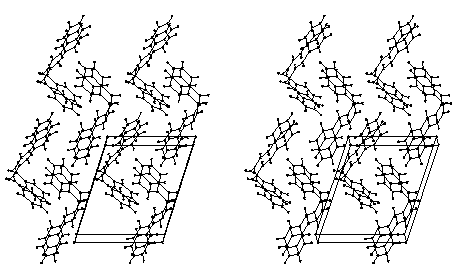
Figure 4. Stereoscopic packing diagram of cis-DBE facing (010) that is inclined by 10° for a better view.
The analysis of the packing arrangements in the Figures 3 and 4 shows that there is not enough free space for the modelling of a cooperative rotational mechanism. Thus rotations in opposite directions at the ends of the double bonds in the fixed lattices become impossible at about ±20° for all different positions in both cases due to excessive interactions. Furthermore, the neighboring molecules do not have much freedom, unlike the situation in the previously analyzed cases.[1] However, inspection of the crystal lattices in Figures 3, 4 indicates, that E/Z-isomerization may be geometrically feasible if the benzoyl groups do not rotate with the double bond C but move approximately in the same plane which they occupy while one of the C-H groups of the double bond turns by 180°. Such a mechanism resembles the so-called "hula twist" movements that have been described for 1,3,5-hexatrienes[6] and is formulated in Scheme 1.
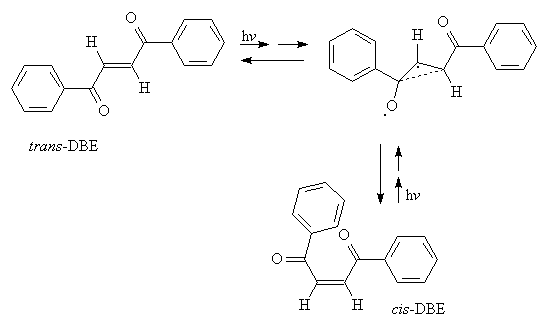
Scheme 1. Presumed E/Z-photomechanism of trans-DBE in the crystal leading directly to the same conformer that is present in the crystals of cis-DBE, however, the sterically feasible back reaction does not occur in the crystal.
The main feature is, that the benzoyl group does practically not leave its plane,
making this mechanism sterically feasible for Figure 3 and for Figure 4, even though the
back photoreaction is not observed in the crystal for other reasons to be discussed below.
Anyhow, there is probably enough energy accumulated after light absorption for the
deactivation via a biradical like multicenter bonded three-membered ring.
It remains to rationalize the unidirectional behavior and the anisotropy in the molecular
movements of the phase rebuilding step in Figure 1b. The packing diagrams give a clue for
the understanding of both observations. It can be nicely seen from Figure 4 that the bent
molecules are so heavily interlocked, that any far-reaching molecular movements are
efficiently impeded. As any alleged trans-DBE molecule, which cannot fit the
lattice due to its totally different shape, could not start such movements, the presumed
photolysis of cis-DBE to give trans-DBE does not work. Further examples for
non-reactivity due to 3D-interlocking have been collected and discussed in Ref.[1b]. On the other hand, Figure 3 shows a layered structure of trans-DBE
crystals. Geometrically totally different cis-DBE does not fit into the lattice and
can thus start the far-reaching molecular movements along these glide planes. It is highly
gratifying to see in Figure 1b, that the direction of the glide planes that are parallel
to (100) corresponds to the direction chosen by the floes that extend in b-direction
along the long crystal axis. The complete preference is lost at higher conversions in the
surface area: exiting material creates holes and submicrocracks that will be used for
further molecular movements. Additionally, when the product crystals form in the phase
transformation, they do not necessarily require the help of the initial lattice and
indeed, the surface between the features has also changed due to photoreaction (see Figure
1).
Conclusions
A single crystal to single crystal reaction[3] has not
been confirmed for the photolysis of trans-DBE. Rather long-range molecular
movements were found that are strictly guided by the crystal packing during the initial
phase rebuilding step. The chemical process must choose the sterically least demanding
option. The nonreactivity of cis-DBE is rationalized by the obvious impossibility
for long-range molecular movements in its crystals. The distinction between rotational and
twist mechanism in solid-state E/Z-photoisomerizations in cases where both appear
feasible will require techniques such as femtosecond spectroscopy, however, the
solid-state mechanisms have to be investigated first. It is to be expected that SNOM
measurements (scanning near-field optical microscopy)[1a]
will again show that features and changed surface have the same chemical composition. Such
experiments will be reported soon.
Literature
[1] a) G. Kaupp, M. Haak, Angew. Chem. 1996, 108, 2948-2951; G.
Kaupp, M. Haak, Angew. Chem. Int. Ed. Engl. 1996, 35, 2774-2777;
b) G. Kaupp, in Comprehensive Supramolecular Chemistry, ed. J.E.D. Davies and J.A.
Ripmeester, Elsevier, Oxford 1996, Vol. 8, p. 381-423 and 21 color plates.
[2] G. Kaupp, Adv. Photochem. 1995, 19, 119-177.
[3] J.C.J. Bart, G.M.J. Schmidt, Recl. Trav. Chim. Pays-Bas, 1978, 97,
231-238.
[4] D. Rabinovich, G.M.J. Schmidt, Z. Shaked, J. Chem. Soc. B, 1970, 17-24.
[5] H. Nakanishi, W. Jones, J.M. Thomas, M.B. Hursthouse, M. Motevalli, J. Chem. Soc.
Chem. Commun., 1980, 611-612; H. Nakanishi, W. Jones, J.M. Thomas, Chem.
Phys. Lett., 1980, 71, 44-48.
[6] A.M. Müller, S. Lochbrunner, W.E. Schmid, W. Fuß, Angew. Chem., 1998, 110,
520-522; A.M. Müller, S. Lochbrunner, W.E. Schmid, W. Fuß, Angew. Chem. Int. Ed.
Engl., 1998, 37, 505-507.