| Papers and Posters | Site Home Page |
EFFECT OF LOW INTENSITY ULTRAVIOLET LIGHT (280 NM)
ON THE ACTIVITY OF PHOTOSYSTEM II PARTICLES
J. A. Ségui, V. Maire, I. S. Gabashvili and M. Fragata
Université du Québec ŕ Trois-Rivičres, Département de Chimie et Biologie, Section de Chimie et Groupe de Recherche en Energie et Information Biomoléculaires (GREIB), Trois-Rivičres, Que, G9A 5H7, Canada
Correspondence: Mário Fragata, fragata@uqtr.uquebec.ca
Keywords:
a-helix, b-strand, extended chain,
FT-IR, infrared, oxygen evolution,
photosystem II, PSII, SDS-PAGE, ultraviolet light
List of abbreviations:
Chl, chlorophyll
D1 or D2, D1 or D2 proteins of PSII-RC
DCBQ, dichlorobenzoquinone
FT-IR, Fourier transform infrared
IR, infrared
OEC, oxygen-evolving complex of PSII
PSII, photosystem II
PSII-RC, reaction center of PSII
P680, reaction center of PSII
QA, quinone QA in PSII
RC, reaction center
SDS-PAGE, sodium dodecylsulfate-polyacrylamide gel electrophoresis
Tyr, tyrosine
UV, ultraviolet
UVB, UV light in the 280-320 nm range
UV280, UV light of 280 nm wavelength
Y, tyrosine
![]()
TABLE OF CONTENTS
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. UV effect on oxygen evolution and protein content in PSII
3.2. FT-IR spectrum of PSII from 1800 to 1500 cm-1
3.3. UV effect on the infrared absorbance in PSII
3.4. UV-induced changes of structure-function correlations in
PSII
4. Concluding Remarks
Acknowledgements
References
![]()
ABSTRACT
A study was undertaken of the effect of ultraviolet radiation of 280 nm wavelength (UV280) and low intensity (2.0 W/m2) on the structure and oxygen-evolving activity of photosystem II (PSII) particles isolated from thylakoid membranes of plant chloroplasts. The results show that an irradiation time of 40 min causes an almost complete loss of oxygen-evolving activity in PSII. The activity remaining after 15, 20, 30 and 40 min is respectively 52, 44, 27 and 12 % of the activity in control PSII particles kept in obscurity. A difference FT-IR spectroscopy study of PSII particles irradiated for a period of time of 30 min with UV280, i.e., [PSII irradiated with UV280]-minus-[PSII non-irradiated] indicates that the UV effect is seen throughout the major regions delimited in the PSII spectrum as significant absorbance changes in the amide I {1696-1620 cm-1) and amide II (1580-1520 cm-1) regions, the tyrosine regions (1620-1580 cm-1; n8a, n8b and n19a vibrational modes), and the chlorophylls region (1750-1696 cm-1). A major conclusion in this study is the demonstratation that the stability of the functional centers in photosystem II is most likely dependent on a overall dynamic equilibrium between the a-helix conformers of the PSII proteins and extended chain (b-strand) structures. It is suggested that this a-helix/b-strand transition may well a means of regulation of the oxygen-evolution complex function.
![]()
1. INTRODUCTION
The decrease in the concentration of the stratospheric ozone has caused the atmosphere to become more transparent to the ultraviolet (UV) radiation, particularly of wavelengths between 280 and 320 nm (UVB) (see, e.g., [1]). As a consequence, the photosynthetic productivity of both terrestrial plants and aquatic ecosystems has been considerably affected [2-4]. This has stimulated numerous investigations on the physiological and biochemical aspects of the ultraviolet effect in plants exposed to radiation in the UVB spectral range (see, e.g., [2,5,6]).
For example, Desai [7] showed that the slow component of the delayed light emission and the photosystem II (PSII) activity in plant chloroplasts are inhibited with UV light of 271 nm wavelength, while on the contrary the PSI activity is only slightly affected. On the other hand, Trebst and Depka [8] found that UV radiation of 254 nm inactivates irreversibly the reaction center of PSII (PSII-RC) owing to the damage caused to the D1 protein. In this same connection, Renger et al. [3] concluded that the manganese cluster (Mn-cluster) in the PSII protein complex is one of the targets of the UV radiation in the neighborhood of the D1 protein. Another interesting study was performed by Melis et al. [9]. These authors observed that UV light of 295, 302 and 335 nm causes the partial degradation of the PSII-RC proteins and the inhibition of the semiquinone anion formation at QA, thus affecting the electron-transport in PSII.
The purpose of the present study is the investigation of the molecular targets of the UVB radiation of 280 nm wavelength, hereunder referred to as UV280, in the PSII complex. For this purpose, we used Fourier transform infrared (FT-IR) spectroscopy to study structural variations produced in PSII particles irradiated with UV280 of low intensity (2.0 W/m2). The expected targets of the UV280 radiation are therefore
(i) the proteins containing aromatic amino acids such as tyrosine
(Tyr or Y) which is present, for instance,
in the D1 and D2 proteins of PSII-RC [10,11]
and in the oxygen evolving complex (OEC)
in PSII (see, e.g., [12,13]),
(ii) the chromophores in the pigment-protein complexes, e.g., the
chlorophylls (Chl) in the photochemical
reaction center P680 of PSII-RC, and
(iii) most probably also the quinone QA in PSII.
Finally, it is worth noting that Melis et al. [9] concluded also that the UVB light does not appear to affect the primary charge separation between Chl and the pheophytin (Phe) in P680. However, the FT-IR study presented below (see also [6]) indicates that the molecular species absorbing at 280 nm, that is, the Chl pigments and the Tyr residues, are targets of the low intensity UV280. We shall discuss whether the observed infrared changes in the FT-IR spectra are ascribable to molecular degradation (see [6]), or, alternatively, to geometrical or conformational changes in the PSII proteins organization capable of explaining the observed loss of oxygen evolution activity.
![]()
2. MATERIALS AND METHODS
Chemicals. Bis-[2-hydroxyethyl]-imino-tris-[hydroxymethyl]methane (bis-Tris), sodium dodecylsulfate, acrylamide, n-dodecyl-b-D-maltoside, dichlorobenzoquinone and tyrosine were purchased from Sigma Chemical Company (St. Louis, MO). All other products were obtained from Fisher Scientific Company (Fair Lawn, NJ).
Isolation of PSII particles and UV irradiation. The PSII membranes were
extracted from leaves of one-week-old barley seedlings grown on vermiculite in a
controlled environment cabinet (continuous white light, 298 K, 10 W/m2) according to the
method of van Leeuwen et al. [14]. After extraction, the isolated PSII particles
were solubilized in a buffer containing 20 mM bis-Tris-HCl (pH 6.5), 20 mM MgCl2, 5 mM
CaCl2, 10 mM MgSO4, 400 mM sucrose and 0.03 % n-dodecyl-beta-D-maltoside. The purity of
this PSII preparation was first checked by SDS-PAGE, then stored at 143 K.
The PSII samples (0.01 to 0.1 mg Chl/mL) were irradiated with light from a 1
kW Xenon lamp, model LH151N from Schoeffel Instruments Corporation (Westwood, NJ). The 280
nm wavelength was isolated with an interference filter from Oriel Corporation (Stratford,
CT). The intensity of the UV280 radiation (2.0-2.7 W/m2) was measured with a
radiometer/photometer , model88XLC, from 3M Photodyne Inc. (Camarillo, CA), equipped with
a radiometric sensor head, model 400. The dark control and UV-irradiated PSII preparations
were incubated under a current of nitrogen in a chamber kept at 277 K to avoid spurious
oxidation and thermal inactivation of the samples.
Oxygen evolution measurement and SDS-PAGE. Oxygen evolution by the PSII
particles was monitored with a Clakck-type electrode (Hansatech D. W. Oxygen Electrode
Unit, King's Lynn, Norfolk, U.K.), with 350 microM dichlorobenzoquinone (DCBQ) as
exogenous electron acceptor and 10-20 microL samples of PSII. The samples were irradiated
with white light at saturation intensity obtained from a light source, model 180, of
Dolan-Jenner (Woburn, MA). Chlorophyll concentration was determined according to the
method of Arnon [15].
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of the
PSII polypeptides and the molecular mass markers (Bio-Rad Laboratories, Richmond, CA) was
performed for 90 minutes at room temperature in a discontinuous system (5 and 12.5 %
acrylamide in the stacking and the resolving gel, respectively) in the presence of 6 M
urea. All other details (cathodic and anodic buffers, staining and destaining solutions)
are given in [6]. The molecular mass of the PSII polypeptides was determined in LKB
Ultroscan XL Enhanced Laser Densitometer (Broma, Sweden).
FT-IR measurements and spectral analyses. First, aliquots of PSII
suspensions were centrifuged at 28700 g for 30 min (277 K) and the pellet was suspended in
3 mL D2O. The pellet was subjected to a second centrifugation and the final pellet was
suspended this time in a D2O volume sufficient to make a solution with a Chl concentration
of 0.333 mg m/L. An aliquot of each sample was then layered on BaF2 plates and let
dehydrate. The stability of the PSII preparations was controlled at each step of the
experimental procedures. To this end, the oxygen-evolution activity of the PSII
preparations was measured before and after the FT-IR measurements. The observed
differences were never greater than about 8%, meaning therefore that the integrity of the
PSII samples was preserved throughout this work.
The tyrosine samples (powder) were layered between NaCl or ZnSe plates. The
samples of amorphous chlorphyll a were prepared as described in [16] and layered in
ZnSe plates.
The infrared measurements were made in a BOMEM FT-IR spectrophotometer, Model
DA 3.2, equipped with a MCT detector (nitrogen cooled) and a KBr beam-splitter, or in a
Perkin-Elmer FT-IR spectrophotometer, Model 2000. In general, 100 interferograms were
collected and co-added and the infrared spectra were obtained upon subtraction of the
spectrum of the plates used for deposition of the samples. Spectral resolution was 2-4
cm-1 (BOMEM) or 1 cm-1 (Perkin-Elmer).
The spectra were processed using the GRAMS/386 program (Galactic Industries
Corporation, Salem, NH). Prior to data processing the FT-IR spectra were corrected for
their content in D2O by a standard subtraction procedure [17]. To obtain the
difference FT-IR spectra, i.e., [PSII irradiated with UV280]-minus-[PSII non-irradiated],
the method developed by Heimburg and Marsh [18] was applied (see also [19]).
In short, the area under the band envelope in the region under study of the [PSII
irradiated with UV280] and [PSII non-rradiated] spectra were first normalized according to
the expression An=kAo, where Ao is the integral of the observed surface under the band
envelope calculated with the Spectra-Calc Program (Galactic Industries Corporation, Salem,
NH), k is a scale factor that normalizes Ao to 100, and An is the normalized area, i.e.,
100. Then, the [PSII irradiated with UV280] - [PSII non-irradiated] subtraction of the
spectra was performed.
![]()
3. RESULTS AND DISCUSSION
3.1. UV effect on oxygen evolution and protein content in PSII
Fig. 1 is a representation of the effect of low
intensity UV280 light (2.0 W/m2) on the oxygen evolution in isolated PSII particles as a
function of the irradiation time. It is noted first that the irradiated particles were
kept at 277 K to avoid the effect of thermal deactivation of the oxygen evolving complex
(OEC). It is seen that an irradiation time of 40 min causes an almost complete loss of
oxygen-evolving activity. In fact, Fig. 1 shows that the activity remaining after
15, 20, 30 and 40 min is respectively 52, 44, 27 and 12 % of the activity in control PSII
particles kept in obscurity at 277 K.
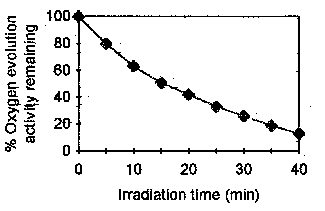
Figure 1.
Oxygen evolution in PSII particles irradiated with UV light of 280 nm wavelength and low intensity (2.0 W/m2). The chlorophyll (Chl) concentration is 20 mg/mL. The average 100 % oxygen evolution is 352 mmol O2/mg Chl/h. The first step undertook in this work to investigate the
mechanisms underlying the effect seen in Fig. 1, was to ascertain whether the loss
of oxygen-evolving activity resulted from a concomitant loss of the PSII proteins most
essential for the OEC function. This experiment is described in Fig. 2. The figure
represents the PSII protein losses observed at 277 K upon the irradiation of the PSII
particles with UV280 light for a period of 90 min. The protein complexes that presented a
measurable variation in concentration were the proximal antenna (PA), the reaction center
of PSII [RC (D1,D2)], and the extrinsic proteins of the oxygen-evolving complex (OEC). It
is seen that the % variations observed after 90 min irradiation are generally smaller than
about 11.4 % (D2 protein of the PSII-RC), and can be as low as 1.6 %.
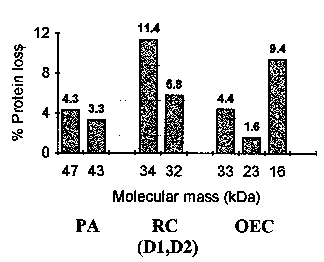
Figure 2.
Protein loss in photosystem II (PSII) irradiated 90 minutes with UV light of 280 nm wavelength and low intensity (2.0 W/m2). PSII protein complexes affected by the UV radiation: D1,D2, D1 and D2 proteins in the PSII reaction center (RC); OEC, oxygen-evolving complex; PA, proximal antenna; RC, reaction center.The interesting point in the afore discussed experiments is that 90 min is an irradiation time about six times larger than the irradiation time required to induce a 50 % loss of oxygen-evolving activity in PSII, i.e., 15 min (cf. Fig. 1). If the data in Fig. 2 are extrapolated to a 15 min period of irradiation with UV280 light, one gets protein losses of less than 2 % for the D2 protein of PSII-RC and as low as 0.3 % for the extrinsic protein of 23 kDa.
A straightforward conclusion is to interpret the large decrease of
PSII activity at irradiation times of less than 30 min not be caused by the observed low
protein loss, but instead by some kind of structural rearrangement induced by the
ultraviolet light of 280 nm wavelength. This is discussed in next section.
3.2. FT-IR spectrum of PSII from 1800 to 1500 cm-1
A typical FT-IR spectrum from 1800 to 1500 cm-1 of the PSII particles used in this work is display in Fig. 3 (A). The decomposition of such spectra into several regions corresponding to the various molecular conformations into which the proteins are folded has been achieved successfully by several authors (see, e.g., [13,20-22]). The major five regions identified between 1800 and 1500 cm-1 in the spectra of PSII are at:
(i) 1750-1696 cm-1: Chlorophylls
(ii) 1696-1620 cm-1: Amide I region
1696-1665 cm-1: turns (e.g., b-turns), antiparallel b-sheet (~1693
cm-1)
1658-1654 cm-1: a-helix
1648-1641 cm-1: random structures,
loops
1640-1620 cm-1: b-sheet
(~1636 cm-1), b-strands (extended chains: ~1626 cm-1)
(iii) 1620-1580 cm-1: Aromatic side chains (e.g., tyrosine: n8a, n8b)
(iv) 1580-1520 cm-1: Amide II region
(v) 1520-1500 cm-1: Tyrosine (n19a)
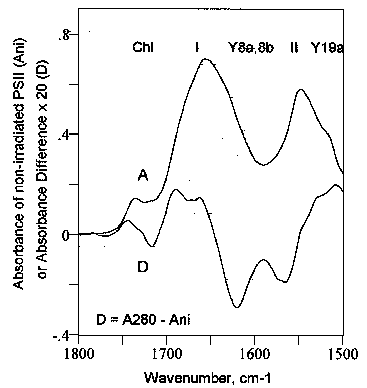
Figure 3.
Infrared spectrum of non-irradiated photosystem II (PSII) particles (A), and difference FT-IR spectrum (D), i.e., [PSII irradiated 30 minutes with UV280)] - [PSII non-irradiated]. SR (spectral regions): Chl, chlorophyll; I, II, amide I and amide II; Y8a,8b, Y19a, n8a, n8b and n19a vibrational modes of tyrosine (Wilson numbering [23]). Briefly, the amide I region (Fig. 3, I) is
associated with the in-plane C=O stretching vibration (~ 80 %) weakly coupled with CN
stretching and CCN deformation, and amide II region (Fig. 3, II) originates
in an out-of-plane combination of in-plane NH bending (~ 60 %) strongly coupled with CN
stretching (~ 40 %). The chlorophylls region (Fig. 3, Chl) contains a
major band at about 1737 cm-1 generally attributed to the ester C=O and the carbomethoxy
group [16,24] which in Chl a are at the the 7c- and 10a-positions (cf. Fig. 4).
The other relevant Chl band observed in near region (~ 1693 cm-1) is the C=O stretching
mode of the free 9-keto group in ring V. This band, however, overlaps with the amide
I band of PSII therefore rendering its analysis somehow difficult; we will come
again to this matter in a forthcoming paper. Finally, the tyrosine regions
are seen from 1620 to 1580 cm-1 [n8a and n8b
vibrational modes (Wilson numbering [23])] (Fig. 3, Y8a,8b), and 1520 to
1500 cm=1 (n19a vibrational mode) (Fig. 3, Y19a).
The n8a and n8b modes are the
splitting components of the ring stretching fundamental vibration in the phenol ring, that
is, the double degenerate e2g no. 8 of Wilson [23], whereas n19a
is assigned to a mixture of ring stretch plus deformation.
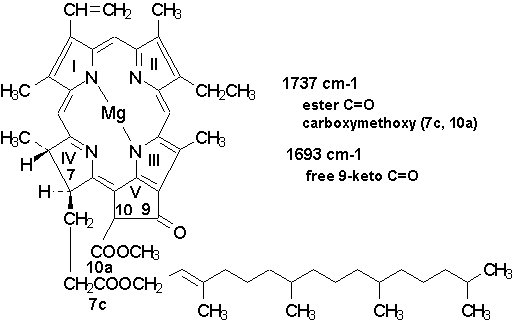
Figure 4. Structural formula of
chlorophyll a with indication of the major infrared transitions expected in the
1750-1690 cm-1 region.
3.3. UV effect on the infrared absorbance in PSII
Fig. 3 displays the difference FT-IR spectra (D) of PSII
particles irradiated at 277 K for a period of time of 30 min with UV light of 280 nm
wavelength, i.e., [PSII irradiated with UV280]-minus-[PSII non-irradiated].
The UV effect is seen throughout the major regions delimited in the PSII spectrum as
significant absorbance changes in the amide I and amide II regions (i), the
tyrosine regions (ii), and the chlorophylls region (iii).
(i) The amide I and amide II regions
On the one hand, the absorbance difference FT-IR spectrum in Fig. 3 (D) shows clearly a UV-induced absorbance increase in the amide I region at 1696-1665 cm-1, i.e., the turns (e.g., b-turns) and antiparallel b-sheet (~1693 cm-1 region), and at 1658-1654 cm-1, that is, the a-helix region. Absorbance maxima are observed at 1693 cm-1 and between 1673 and 1657 cm-1. In contrast, the UV280 radiation causes an absorbance decrease in the spectral region between 1640 and 1620 cm-1, that is, the b-sheet (~1636 cm-1) and b-strands (extended chains: ~1626 cm-1) region. A minimum is observed at about 1621 cm-1.
On the other hand, Fig. 3 displays an infrared absorbance loss in the amide II region with spectral minima between 1572 and 1566 cm-1, thereby indicating changes occurring in the framework of the proteins skeleton (see above).
In brief, a most interesting observation is first the infrared
absorbance increase at about 1657 cm-1, meaning that the UV280 effect is primarily an
augmentation of a-helix structures in PSII. Moreover,
this increase in the helical content in PSII is concomitant with the increase of
antiparallel b-sheet structures. Secondly, on the
basis of the work by Arrondo et al. [25] and De Las Rivas and Barber [22]
the absorbance decrease observed at 1626 cm-1 is attributed with great certainty to loss
of extended chain structures (i.e., b-strands) in PSII.
In this respect, the assignment of the 1626 cm-1 band to protein chains in extended chain
configurations is particularly interesting in the study of structure-function in
macromolecules since the b-strands are structures that are
often found in hydrogen-bonding networks, that is, molecular arrangements formed of
hydrogen-bonding donors and acceptors in amino acid residues which do not participate in
intramolecular b-sheets. These molecular patterns are for
the most part involved in hydrogen-bonding interactions with other molecular
structures. However, one has to acknowledge that an infrared absorbance
decrease is as well seen around 1636 cm-1; that is to say, in a spectral region
related to transitions arising from intramolecular carbonyl vibrations of b-sheets.
(ii) The tyrosine regions
In the 1622-1580 cm-1 region, the difference FT-IR spectrum represented in Fig.
3 (D) displays two minima. The most prominent is at about 1620 cm-1 and
the less pronounced at about 1590 cm-1. Now, the 1620-1580 cm-1 region is
particularly interesting in the infrared study of proteins on account of its sensitivity
to molecular perturbations (ring deformations, symmetry losses) originating in intra- and
intermolecular interactions. The spectral data in Fig. 3 (D) is
therefore a reasonable indication that the configuration of the tyrosine ring in the PSII
proteins has been affected to a considerable extent.
The expected affected modes of tyrosine are n8a
and n8b which are the splitting components of the ring
stretching fundamental vibration in pheol ring, i.e., the double degenerate e2g no. 8 of
Wilson []. The corresponding frequencies are usually seen around 1605 (n8a) and 1580 cm-1 (n8b) with a wavenumber
dispersion that can be as high as 30 cm-1 in different aromatic compounds (see, e.g., [26,27]).
It is worth noting that in spite of the fact that the deperturbed n8
fundamental in benzene is active in Raman at approximately 1600 cm-1 [23,28], this
transition is not expected, from symmetry considerations, to be observed in infrared (see
discussions in [23]). However, the 1600 cm-1 transition is observed in
splitted form in most benzene derivatives where it usually displays a strong
intensity. This is seen, for example, in the phenol spectrum which exhibits two main
bands with maxima at 1606 and 1596 cm-1 identified as the n8a
and the n8b splitting modes [13]. In tyrosine the
expected frequencies of these vibrational modes are usually observed around 1608 (n8a) and 1590 cm-1 (n8b) (see, e.g., [13]).
This is in good agreement with the infrared data in the difference FT-IT spectrum given in
Fig. 3 (D).
In the 1520-1500 cm-1 region, the spectral region from 1520 to 1500 cm-1 results
from a tyrosine vibration attributed to the high frequency splitting mode of the benzene
fundamental vibration no. 19 of Wilson, i.e., n19a, which is
assigned to a mixture of ring stretch plus deformation [23]. Fig. 3 (D)
shows that the major changes are an absorbance increase with a maximum at about 1511 cm-1
which is assigned to the n19a vibrational mode.
(iii) The chlorophylls region
Fig. 5 displays the difference FT-IR spectrum of PSII in the chlorophylls region between 1760 and 1700 cm-1. It is seen that the UV280 light caused an infrared absorbance increase at about 1744 cm-1, and a concomitant absorbance loss around 1718 cm-1. This is interesting since usually the transitions identified in the 1760-1700 cm-1 region are a band at about 1737 cm-1 assigned to the ester C=O and the carbomethoxy group (see section 3.2, and Fig. 4), and the band observed near ~ 1693 cm-1 which is the C=O stretching mode of the free 9-keto group in ring V (see, e.g., [16,24]).
To interpret the difference FT-IR spectrum in Fig. 5 one
may consider either (a) a UV-induced change in the molecular environment of the
chlorophyll molecules in the pigment-protein complexes of PSII, or, alternatively, (b)
a photodegradation of the pigments as the result of irradiation of the PSII preparations
with UV light of 280 nm wavelength (see Ségui and Fragata [6]). However, the
chlorophyll degradation observed in [6] at 677 nm (QY(0,0)
transition of Chl a; see [29]) was about 10 % of the total chlorophyll
content for an irradiation time of 90 min; that is, much longer than the 30 min
irradiation used in the experiments described here. At 624 nm, i.e., the QX(1,0) transition of Chl a, the observed
UV-induced photodegradation was lower than 7 % of the total chlorophyll content upon an
irradiation period of also for a 90 min [6]. The corresponding chlorophyll
degradation for an irradiation period of about 30 min were calculated to be respectively ~
3 % (677 nm) and ~ 2 % (624 nm).
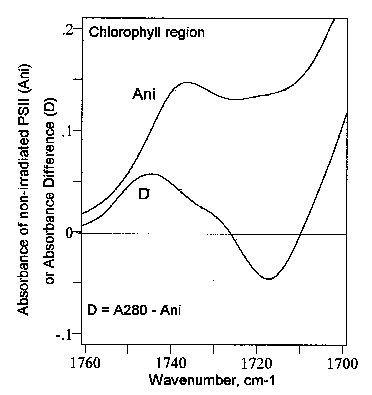
Figure 5.
Difference spectrum of photosystem II (PSII) in the chlorophylls region from 1800 to1700 cm-1. Ani, absorbance spectrum of non-irradiated PSII. D, difference FT-IR spectrum of PSII, i.e., [PSII irradiated with UV280]-minus-[PSII non-irradiated]. A280, PSII irradiated 30 min with 280 nm light. On the whole, the afore discussed data are a clear indication that
after an irradiation period of 15 to 30 min with UV280 light one may expect that the
molecular perturbations causing the loss of activity in the PSII particles are not
necessarily protein degradations, but instead changes affecting their three-dimensional
structures. Moreover, we hypothesize as a first approximation that these structural
perturbations are changes affecting the PSII proteins either globally or, more likely,
only at well-delimited regions in the supramolecular PSII complex, e.g., changes in
specific protein domains or motifs.
3.4. UV-induced changes of structure-function correlations in PSII
A major finding in this work is the observation that the UV-induced increase in the a-helix content in the PSII complex is accompanied by an opposite variation in the extended chains (b-strands) content [cf. Fig. 3 (D)]. In this framework, it is obvious that the PSII structures might evolve to a to three-dimensional conformations characterized by higher hydrogen-bonding content. If this is the case one may be able to detect an increase of bound-OH groups in the infrared spectrum of PSII particles irradiated with light of 280 nm wavelength. This is shown in Fig. 6.
In short, Fig. 6 represents the FT-IR spectrum of non-irradiated PSII particles (Ani) and the difference FT-IR spectrum (D) of PSII particles irradiated with light of 280 nm wavelength. The spectral region around 3548 cm-1 is assigned to free OH groups, whereas the region around 3305 cm-1 corresponds to bound OH groups (see, e.g., [16]. The difference FT-IR spectrum (D) indicates clearly that irradiation of the PSII particles with UV280 light causes a marked decrease in the concentration of free OH groups which is accompanied by an almost identical increase in the concentration of bound OH groups. This corroborates the observation discussed above of an increase in the a-helix content in PSII accompanied by a decrease in the extended chains (b-strand) content (see Fig. 3 and section 3.3i).
A second question that emerges from this study is that PSII
contentsd in b-sheets and antiparalell b-sheets vary as well in opposite senses upon
irradiation of the PSII particles with UV280 light. It is possible that this effect
could be also explained by a free OH/bound OH equilibrium as discussed above for
the a-helix/b-strand transition.
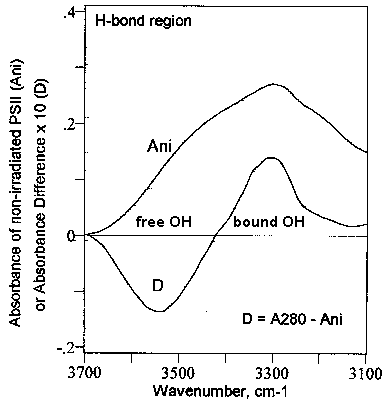
Figure 6. Difference spectrum of
photosystem II (PSII) in the hydrogen-bond region. Ani, absorbance spectrum
of non-irradiated PSII. D, difference FT-IR spectrum of PSII, i.e., [PSII
irradiated with UV280]-minus-[PSII non-irradiated]. A280,
PSII irradiated 30 min with 280 nm light.
A corollary from the above discussed considerations is that the PSII proteins conformations capable of assuring a maximum number of functional oxygen-evolving centers would seem to be intimately dependent on a dynamic equilibrium between the a-helix and the extended chain (b-strand). In absence of UV280 irradiation which induces an extreme loss of functional centers, one observes respectively a lower a-helix and higher b-strand content than in PSII preparations irradiated with UV280 light. It has been suggested before [30] that a similar type of spatio-temporal fluctuations in the PSII supramolecular complex may occur in PSII particles subjected to temperatures of the order of 313 K, or higher {see discussions in [30]). Such a-helix/b-strand transitions may give rise to simple background noise effects in the PSII function, or to more important functional deviations. Most importantly, it is also plausible that these changes in the secondary structures of the PSII proteins may constitute an efficient means of control of the PSII activity in the thylakoid membrane.
![]()
4. CONCLUDING REMARKS
In conclusion, it is interesting to remark that the structural changes occurring in the course of protein folding and unfolding provide useful information about the intra- and intermolecular interactions between specific amino acid residues and/or protein domains that are essential to maintain the physiologically active macromolecular conformations (see discussions in [31]). Such information is fundamental for the comprehension of the molecular arrangements or local structure order that are involved directly or indirectly in biological catalysis. This has been the scope of a wide range of investigations on the function of the thylakoid membrane proteins. The present study (see also [30]) is a novel step in this direction as it demonstrates that the stability of the functional centers in photosystem II is most likely dependent on a overall dynamic equilibrium between the a-helix conformers of the PSII proteins and extended chain (b-strand) structures. The study of these questions is being pursued now with new experimental procedures. We intend, in addition, to undertake the dynamic modeling of the a-helix/b-strand transition in computer reconstructed PSII and/or thylakoid membranes.
![]()
ACKNOWLEDGEMENTS
This work was supported by grant OGP0006357 from the N.S.E.R.C. Canada and
institutional grants from the Université du Québec ŕ Trois-Rivičres (UQTR) to MF. We
thank Drs. C. Daneault and S. Robert for making their Perkin-Elmer FT-IR spectrometer
available to our studies. We are also very grateful to Jimmy Prasakis and Carl Pedneault
for assistance with the FT-IR spectrometer and the computer facilities. We wish as well to
thank the staff of the Service d'Informatique and the Service d'Audio-Visuel of the UQTR
for their precious help and advice in this occasion, and in many other occasions.
![]()
REFERENCES
[ 1] McFarland, M. and Kaye, J. (1992) Photochem. Photobiol. 55,
911.
[ 2] Bornman, J. F. (1989) J. Photochem. Photobiol. B: Biol. 4,
145.
[ 3] Renger, G., Voelker, M., Eckert, H. J., Fromme, R., Hohm-Veit, S. and
Graeber, P. (1989)
Photochem. Photobiol. 9,
97.
[ 4] Gala, W. R. and Giesy, J. P. (1991) Ecotoxicol. Environ. Safety 22,
345.
[ 5] Panagopoulos, I., Bornman, G. F. and Bjorn L. O. (1990) J. Photochem.
Photobiol. B: Biol. 8, 73.
[ 6] Ségui, J. and Fragata, M. (1994) Plant Physiol. (Life Sci. Adv.) 13,
321.
[ 7] Desai, T. S. (1990) Photosynth. Res. 25, 17.
[ 8] Trebst, A. and Depka, B. (1990) Z. Naturforsch. 45c, 765.
[ 9] Melis, A., Nemson, J. A. and Harrison, M. A. (1992) Biochim. Biophys.
Acta 1100, 312.
[10] Barry, B. and Babcock, G. T. (1987) Proc. Natl. Acad. Sci. USA 84,
7099.
[11] Svensson, B., Vass, I., Cedergren, E. and Styring, S. (1990) EMBO J. 9,
2051.
[12] Xu, Q., Nelson, J. and Bricker, T. M. (1994) Biochim. Biophys. Acta 1188,
427.
[13] Gabashvili, I. S., Menikh, A., Ségui, J. A. and Fragata, M. (1998) J. Mol.
Struct. 444, 123.
[14] van Leeuwen, P. J., Nieveen, M. C., van de Meent, J., Dekker, J. P. and van
Gorkom, H. J. (1991)
Photosynth. Res. 28, 149.
[15] Arnon, D. I. (1949) Plant Physiol. 14, 1.
[16] Chapados, C., Lemieux, S. and Carpentier, R. (1991) Biophys. Chem. 39,
225.
[17] Ahmed, A. and Tajmir-Riahi, H.-A. (1994) J. Mol. Struct. 319,
145.
[18] Heimburg, T. and Marsh, D. (1993) Biophys. J. 65, 2408.
[19] Lee, D. C., Haris, P. I., Chapman, D. and Mitchell, R. C. (1990) Biochemistry
29, 9185.
[20] Krimm, S. and Bandekar, J. (1986) Adv. Protein Chem. 38, 181.
[21] Arrondo, J. L. R., Muga, A., Castresana, J. and Gońi, F. M. (1993) Prog.
Biophys. Mol. Biol.
59, 23.
[22] De Las Rivas, J. and Barber, J. (1997) Biochemistry 36, 8897.
[23] Wilson, Jr., E. B, Decius, J. C. and Cross, P. C. (1955) The Theory of
Infrared and Raman
Vibrational Spectra, McGraw-Hill, New
York.
[24] Fragata, M., Nénonéné, E. K., Maire, V. and Gabashvili, I.
S. (1997) J. Mol. Struct. 405, 151.
[25] Arrondo, J. L. R., Castresana, J., Valpuesta, J. M. and Gońi, F. M. (1994) Biochemistry
33, 11650.
[26] Colthup, N. B., Daly, L. H. and Wiberley, S. E. (1990) Introduction to
Infrared and Raman
Spectroscopy, 3rd ed., Academic Press,
Boston.
[27] Dollish, F. R., Fately, W. G. and Bentley, F. F. (1973) Characteristic Raman
Frequencies of Organic
Compounds. John Wiley, New York.
[28] Wilson, Jr., E. B. (1934) Phys. Rev. 45, 706.
[29] Nordčn, B., Fragata, M. and Kurucsev, T. (1992) Aust. J. Chem. 45,
1559.
[30] Joshi, M. and Fragata, M. (1999) Z. Naturforsch. 54c, 35.
[31] Mombelli, E., Afshar, M., Fusi, P., MariaNI, m., Tortora, P., Connelly, J. P.
and Lange, R. (1997)
Biochemistry 36, 8733.
![]()
THE END

Mário Fragata
Trois-Rivičres, June 13, 1999
![]()