| Papers and Posters | Site Home Page |
ELECTROSPRAY-MASS SPECTROMETRY CHARACTERIZATION AND MEASUREMENT OF
FAR-UV INDUCED THYMINE PHOTOPRODUCTS
Thierry Douki, Magali Court, Jean Cadet
Laboratoire des Lésions des Acides Nucléiques; Service de Chimie
Inorganique et Biologique; Département de Recherche Fondamentale sur la Matičre
Condensée; CEA/Grenoble
17 Avenue des Martyrs; 38054 Grenoble Cedex 9, France
email address: douki@drfmc.ceng.cea.fr
Abstract
Far-UV induced formation of dimeric pyrimidine photoproducts within DNA is a major
cause of the carcinogenic effects of solar light. Determination of the chemical structure
of this class of lesion has been mostly achieved by studies on model compounds. The
present work was aimed at providing mass spectrometry data on the thymine-thymine
photoproducts, including the diastereoisomers of the cyclobutane dimer, the (6-4) adduct,
the related Dewar valence isomer and the spore photoproduct. Fragmentation mass spectra of
the modified bases, nucleosides, dinucleoside monophosphates and dinucleotides were
recorded following electrospray ionization with either triple quadrupolar or ion trap
detection. The results show differences in fragmentation pattern between the different
types of photoproducts. In addition, a drastic effect of the diastereoisomery was observed
for the cyclobutane dimers. Then, a sensitive detection technique was developed for the
analysis of dinucleoside monophosphate photoproducts by high performance liquid
chromatography associated with mass spectrometry in the negative mode with multiple
reaction monitoring detection.
1. Introduction
The mutagenic and carcinogenic effects of solar light are mainly mediated by the UV
portion of its spectrum [1]. In particular, UVB radiation is highly lethal and mutagenic
to cells. At these wavelengths, the mutagenicity and lethality action spectra are
identical to the DNA absorption spectrum. This strongly supports the involvement of
photochemical reactions induced by the direct absorption of UVB light by DNA components.
The main class of UVB-induced lesions consists in dimeric pyrimidine photoproducts (Fig.
1). Efforts have been devoted in the last four decades to elucidate the chemical nature of
these photolesions [2,3]. These include cyclobutane type dimers arising from the
cycloaddition of the C5-C6 double bond of two adjacent pyrimidines, and the pyrimidine
(6-4) pyrimidone adducts which result from the addition of the C5-C6 double of the 5'-end
pyrimidine to the C4 carbonyl or imine group of the 3'-end pyrimidine. Subsequently, the
(6-4) photoproducts can photoisomerize into their Dewar valence isomers upon UVB
irradiation. It can be added that, in dry materials, an additional photoproduct, known as
the spore photoproduct [4], is generated through the addition of the methyl group of a
thymine to the C5 position of an adjacent thymine.
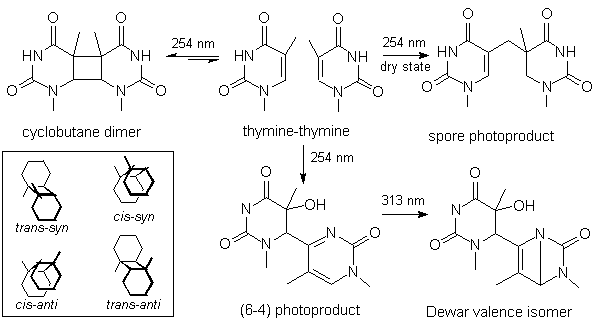
Figure 1: Structure of the thymine
photoproducts studied in this work. The inset shows the different diastereoisomers of the
cyclobutane dimer.
These photoproducts have been studied using various model compounds including bases,
nucleosides and dinucleoside monophosphates [5-17]. Extensive characterization have been
carried out by UV absorption, fluorescence, NMR and mass spectrometry measurements. The
latter technique has mostly been used to determine the molecular weight of isolated
photoproducts but only few extensive studies of the fragmentation patterns have been
reported [18-21]. In the present work, electrospray/tandem mass spectrometry (ES/MS-MS)
was applied to the characterization of thymine-thymine photoproducts at the base,
nucleoside, dinucleoside monophosphate and dinucleotide level. Analyses were carried out
on both a triple quadrupolar spectrometer and an ion trap apparatus. In addition, the
coupling of high performance liquid chromatography to a triple quadrupolar mass
spectrometer used in the multiple reaction monitoring mode (MRM) was developed for the
sensitive detection of thymidylyl-(3'-5')-thymidine (TpT) photoproducts.
2. Material and Methods
2.1 Chemicals
Thymine was purchased from Sigma (St Louis, MO). Thymidine was obtained from
Pharma-Waldhof. Ammonium formate, acetophenone and formic acid were purchased from Aldrich
(Milwaukee, WI). TpT and pTpT were prepared by using a phosphotriester method. Water was
purified on a Milli Q system (Millipore, Molsheim, France).
2.2 Synthesis of the cyclobutane dimers
Thymidine (20 mg) was irradiated in a quartz vial in 30 ml of a 10 mM aqueous solution
of acetophenone. UVB light (300 nm) was provided by 16 lamps of a Rayonet photoreactor
(The Southern New EnglandUltraviolet Company, Handem, MA). Then, the volume was reduced to
2 ml under vacuum. The six diastereoisomers of the thymidine cyclobutane dimers were
separated by successive HPLC purifications [22] with a UV spectrometer set at 230 nm as
the detector. A first separation was performed on a Inertsil ODS 2 octadecylsilyl silica
gel column (250x4 mm ID, 5 µm particle size, Interchim, Montluçon, France) with a
[90:10] mixture of a 25 mM aqueous solution of ammonium formate and methanol used as the
isocratic eluent. The cis-syn and trans-anti diastereoisomers were isolated
as pure material (retention time 9.6 and 25.6 min, respectively). Two fractions that
contained mixture of the (-) trans-syn and the (-) cis-anti on one hand, and
the (+) trans-syn and the (+) cis-anti diastereoisomers on the other hand
were collected at retention times of 11.2 and 12 min, respectively. The two latter
fractions were concentrated under vacuum and injected on a Hypersil NH2 amino
column (250x4.6 mm ID, 5 µm particle size, Interchim, Montluçon, France) with a [7:93]
mixture of a 25 mM aqueous solution of ammonium formate and acetonitrile used as the
isocratic eluent. The retention times of the thymidine cyclobutane dimers were the
following: (-) trans-syn: 8 min, (-) cis-anti: 11.2 min, (+) trans-syn:
8.8 min and (+) cis-anti : 12 min. Diastereoisomers of thymine cyclobutane dimers
were prepared by 88% formic acid hydrolysis (140°C, 2 h) of the corresponding purified
thymidine photoproducts. Formic acid was evaporated under vacuum. The sample was then
solubilized in water. TpT and pTpT cyclobutane dimers were prepared by photosensitization
of 5 mg of the starting material in 20 ml of a 10 mM aqueous solution of acetophenone
using the procedure used for the synthesis of thymidine dimers. The photoproducts were
isolated by reverse phase HPLC as reported [23]. Identification of the photoproducts was
made by comparison of their retention times and UV spectra with those of authentic
standards.
2.3 Synthesis of the (6-4) photoproducts and their related Dewar valence isomers.
The (6-4) photoproduct of thymine was obtained by irradiation of the base in frozen
aqueous solution (20 mg in 20 ml solution). The sample was placed in a 20 cm diameter
petri dish and subsequently frozen on dry ice. It was then exposed to the 254 nm light
emitted by a germicidal lamp for 2 h. Then, the solution was concentrated to 5 ml,
centrifuged and the liquid phase injected on a Inertsil ODS2 octadecylsilyl silica gel
column (250x4.6 mm ID, 5 µm particle size, Interchim, Montluçon, France). The gradient
of elution consisted in a mixture of a 25 mM aqueous solution and acetonitrile. The
proportion of the latter solvent linearly increased from 0 to 5% within 15 min. The
elution was monitored at 315 nm. The retention time of the thymine (6-4) photoproduct was
11.1 min. The 2 diastereoisomers of the thymidine (6-4) adducts were prepared by
irradiation at room temperature of a saturated aqueous solution of thymidine with the 254
nm light emitted by a germicidal lamp. Then, the solution was freeze dried overnight and
the resulting residue was dissolved in a [90:10] mixture of acetonitrile and water. The
two diastereoisomers were then isolated on a semi-preparative Hypersil NH2
amino column (250x10 mm ID, 5 µm particle size) with the UV detector set at 320 nm. The
retention time of the two diastereoisomers was 24 and 32 min, respectively, when a [90:10]
mixture of acetonitrile and 25 mM aqueous solution of ammonium formate was used as the
isocratic eluent. The (6-4) photoproducts of TpT and pTpT were obtained by exposure to 254
nm light for 2 hours of 5 mg of starting material in aqueous solution (20 ml) placed in a
20 cm diameter petri dish. The solution was then concentrated under vacuum and the
photoproducts isolated by reverse phase HPLC as previously reported [13,16]. The Dewar
valence isomers of thymine, thymidine, TpT and pTpT were all prepared using the same
protocol. A 1 ml solution of the corresponding (6-4) photoproduct was placed in a UV
spectrophotometer cell and the UV absorbance at 320 nm was measured. Then, the quartz cell
was placed in a Rayonet photoreactor equipped with sixteen 300 nm lamps. After increasing
times of irradiation, the 320 nm absorbance of the solution was measured. The irradiation
was stopped when no residual 320 nm irradiation remained.
2.4 Synthesis of the spore photoproducts
Three methods were used for the preparation of the thymidine spore photoproducts. i)
Thymidine (100 mg) was solubilized in 50 ml of ethanol. The resulting solution was placed
in a 4x30 cm vial and evaporated overnight. Then, the dry residue was irradiated for 24
hours with a 2x15 W germicidal lamp. A hair drier was blown on the dry film during the all
irradiation to prevent moisture absorption. In a subsequent step, 5 ml of water was added
to the solid film. The resulting suspension was centrifuged and the liquid phase
collected. ii) Thymidine (100 mg) was solubilized in 50 ml of ethanol together with 50 mg
of benzophenone. The resulting solution was placed in 4x30 cm vial and evaporated
overnight. Then, the dry residue was irradiated for 17 hours in a Rayonet photoreactor
equipped with ten 350 nm lamps. Then, 5 ml of water was added to the solid film. The
resulting suspension was centrifuged and the liquid phase collected. iii) Thymidine (300
mg) was solubilized in 50 ml of water. The solution was frozen on dry ice in a 12 cm
diameter petri dish and then irradiated for 2 h with a 254 nm germicidal lamp. The
solution was thawed and irradiated for 30 min in a Rayonet photoreactoer equipped with ten
300 nm lamps in a 150 ml quartz vial. The latter step was aimed at converting (6-4)
photoproducts into their Dewar valence isomers in order to facilitate the subsequent HPLC
purification. The solution was then concentrated to 5 ml under vacuum. Following any of
the three procedures described above, the thymidine spore photoproducts were purified by
reverse phase HPLC on a Uptisphere ODSB octadecylsilyl silica gel column (250x4.6 mm ID, 5
µm particle size, Interchim, Montluçon, France). The isocratic eluent was a [85:15]
mixture of a 25 mM aqueous solution of ammonium formate and methanol used at a flow rate
of 1 ml/min. The retention times of the two diastereoisomers of the thymidine spore
photoproduct were 18 and 23 min. The two first syntheses provided only the slowly eluting
isomer. The two diastereoisomers were characterized by comparison of their UV absorption
and 1H NMR features with published data [2,3]. The thymine spore photoproduct
was obtained by hot formic acid hydrolysis of the related thymidine derivatives.
2.5 ES/MS-MS analyses
All photoproducts were analyzed by electrospray mass spectrometry in the positive mode.
For this purpose, the products were solubilized in a [1:1] mixture of acetonitrile and
water that contained 0.1% formic acid. The photoproducts of TpT and pTpT were also
analyzed in the negative mode in a [1:1] mixture of acetonitrile and water that contained
25 mM ammonium formate. All spectra were recorded with a unity resolution. The same
ionization and fragmentation conditions were used within an homolog series of
photoproducts. Fragmentation mass spectra on a triple quadrupole system were recorded on a
Quattro II spectrometer (Micromass, UK). The argon pressure in the collision cell was set
between 5x10-4 and 10-3 mTorr. Simultaneous acquisition of spectra
with different setting of the collision energy was performed. Mass spectra were also
recorded on an ion trap LCQ spectrometer (Finningan Mat, San Jose, CA). MS2 and
MS3 spectra were recorded for all compounds on the latter apparatus . When the
intensity of the signal was high enough, MSn (n<5) analyses was also carried
out. Detection of TpT photoproducts by HPLC coupled to tandem mass spectrometry was
performed on a API 3000 spectrometer (Perkin-Elmer/Sciex, Toronto, Canada). The HPLC
system consisted of a 7100 Hitachi-Merck pump (Merck, Darmstadt, Germany) associated to a
SIL-9 automatic injector (Shimadzu, Tokyo, Japan). The column was an Uptisphere ODSB
(150x2 mm ID, 5 µm particle size) octadecylsilyl silica gel column (Interchim,
Montluçon, France). The mobile phase was a gradient of 5 mM ammonium formate and methanol
with a flow rate of 200 µL/min. The proportion of methanol rose from 0 to 2 % within 8
min and reached 28 % after 20 min. Methanol was also added at the outlet of the column and
prior to the inlet of the mass spectrometer at a flow rate of 0.2 ml/min. The spectrometer
was operated in the negative mode and the analyses was performed in the MRM mode. Two
transitions were monitored: 545-447 and 545-532. The dwell time was set at 1 s for both
signals.
3. Results and discussion
3.1 Preparation of the photoproducts
The synthesis and purification of the cyclobutane dimers and the (6-4) adducts of thymine, thymidine, TpT and pTpT were achieved using published procedures [7,13,23-25]. The preparation of the Dewar valence isomers by photoisomerization of solutions of (6-4) photoproducts allowed a comparison of the influence of the structure of the adduct (base, nucleoside or dinucleoside monophosphate) on the efficiency of the photoisomerization reaction. The main effect was observed for the thymine (6-4) photoproduct which was found to be converted significantly more slowly than the related thymidine, TpT and pTpT adducts. This reflects a significant change in the photochemical properties of this class of photoproduct when a free rotation along the C6-C4 bond is possible. A much higher fluorescence quantum yield has already been reported for the free base (6-4) adduct with respect to the related modified dinucleoside monophosphate [26].
Emphasis was also placed on the mechanism of formation of the spore photoproduct. The
latter compound has been reported to be obtained from thymidine upon UVC irradiation
either in the dry state or in frozen solution [9]. Interestingly, only one of the two
possible diastereoisomers was obtained when thymidine wass irradiated as a dry film
whereas the two photoproducts were obtained in frozen solution. This clearly shows the
importance of the stacking of the nucleoside in the stereochemistry of the photoreaction.
Two mechanisms have been proposed for the formation of the spore photoproduct [2]. The
first one involves the recombination of the photogenerated 5-(uracyl)methyl and the
5,6-dihydrothymin-5-yl radicals. A concerted mechanism has also been proposed and has been
shown to actually take place in the frozen state on the basis of experiments involving
irradiation of methyl deuterated thymidine [2]. The present photosensitization experiment
of thymidine by benzophenone in the dry state strongly suggests that a concerted mechanism
is also involved in the formation of the spore photoproduct in dry films of thymidine.
Indeed, the energy of the triplet state of benzophenone is close to that of thymine [27].
Therefore, triplet energy transfer from excited benzophenone to thymine is an efficient
process. This has been widely used for the preparation of cyclobutane dimers in aqueous
solution of bases, nucleosides, dinucleoside monophosphates and DNA [16,24,27]. The
observation of the formation of the spore photoproduct upon irradiation of thymidine in
the dry state shows the involvement of the triplet state of the nucleoside, and rules out
the radical mechanism. It is worth mentioning that irradiation of TpT in the dry state did
not allow the isolation of the related spore photoproduct. A likely explanation might be
the presence of water bound to the phosphodiester group. Indeed, humidity has been shown
to be a key parameter in the formation of the spore photoproduct within DNA films [28].
3.2 Mass spectra of the photoproducts of bases and nucleosides
The syntheses described above provided 7 thymine-thymine and 12 thymidine-thymidine
photoproducts. It should be reminded that, for a given series, all types of photoproducts
exhibit the same molecular weight which is 252 and 484, respectively. It is therefore of
interest to determine whether the fragmentation pattern of each individual compound allows
to differentiate the different photoproducts. The interpretation of data presented below
will be based on fragmentation mass spectra obtained on a quadrupolar spectrometer. The
data provided by the analyses carried out on the ion trap system will be discussed when
they provide additional insight in the understanding of the fragmentation pattern of the
various photoproducts. However, it must be emphasized that both type of mass spectra
exhibited the same ions for a given product.
All thymine photoproducts were analyzed by ES/MS-MS (Fig. 2). The main fragment was
observed at m/z=235 for the (6-4) photoproduct of thymine which corresponds to the
dehydration of the C5-C6 bond of the 5'-end thymine. The same fragmentation was obtained
for the Dewar valence isomer. In contrast, the spore photoproduct underwent the loss of 17
mass units. No explanation could be found for the loss of 17 uma from the spore
photoproducts. To rule out a poor calibration of the spectrometer, a mixture of the (6-4)
adduct and the spore photoproduct was analyzed and two distinct ions were observed at m/z=
235 and 236.
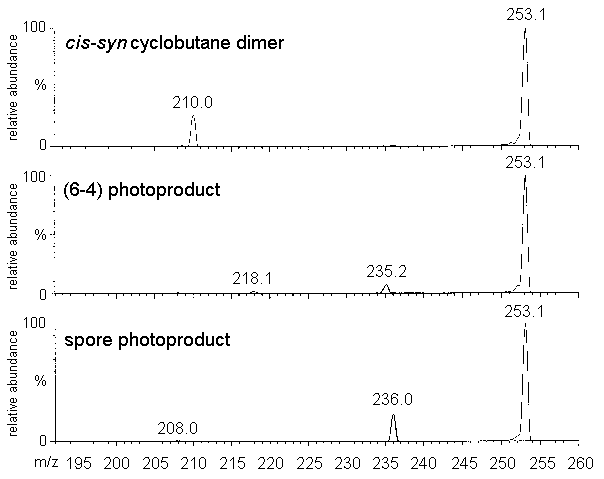
Figure 2. Mass spectra of thymine-thymine
photoproducts. All spectra were recorded under the same conditions on a triple quadrupole
Quattro II spectrometer. The argon pressure in the collision cell was set to 3x10-4
mTorr and the collision energy to 20 V.
The fragmentation pattern of the cyclobutane dimers was found to depend on the
respective orientation of the two C5-C6 bonds (Table 1). Indeed, the spectra of the cis-syn
and the trans-syn isomers were very similar and exhibited a main ion at m/z= 210.
This corresponds to the loss of 43 uma which can be rationalized in terms of the loss of a
HCNO fragment from the pyrimidine ring. This results is in agreement with those obtained
by using laser desorption Fourier transform mass spectrometry. Indeed, the loss of a HCNO
fragment from the [M-H]- pseudo-molecular ion of the cis-syn
cyclobutane dimer was observed [18-21]. In contrast to the observation made on the syn
diastereoisomers, the main ion observed on the ES/MS-MS fragmentation spectrum of the cis-anti
and trans-anti cyclobutane dimers was at m/z=127. The latter ion corresponds to
protonated thymine. This is indicative of a lower stability of the cyclobutane ring of the
anti cyclobutadithymine with respect to that of the syn diastereoisomers.
ion |
cis-syn |
trans-syn |
cis-anti |
trans-anti |
253 |
35 |
76 |
15 |
21 |
235 |
n.d. |
3 |
4 |
5 |
210 |
100 |
100 |
11 |
12 |
127 |
n.d. |
n.d. |
100 |
100 |
Table 1: Relative abundance of the main ions
observed on the mass spectra of thymine cyclobutane dimers. The analyses were performed on
a triple quadrupolar Quattro II spectrometer. The argon pressure in the collision cell was
9x10-4 mTorr and the collision energy was 25 V.
Fragmentation spectra of the diastereoisomeric spore photoproducts of thymidine
exhibited two main ions (m/z=369 and 253 uma) corresponding to the successive loss of the
2-deoxyribose units ([MH-116]+ and [MH-232]+). The interpretation of
the loss of 232 uma as the fragmentation of the two N-glycosidic bonds was
confirmed by ion trap analysis. Indeed, fragmentation of the [MH-116]+ ion
yielded the [MH-232]+ ion. No significant differences were observed between the
spectra of the two diastereoisomers of the thymidine spore photoproducts. The ions at m/z=
369 and m/z=253 were also observed on the MS/MS spectra of the two (6-4) photoproducts,
together with their dehydration products. Analysis with the ion trap spectrometer showed
that the loss of a 2-deoxyribose was favored since it is the only fragmentation observed
on the MS2 spectrum. The loss of one molecule of water is observed on the MS3
spectrum of the [MH-116]+ ion, together the loss of the second 2-deoxyribose
unit and a second molecule of water. A similar fragmentation pattern was obtained for the
related Dewar valence isomers.
The loss of the 2-deoxyribose unit was also the major fragmentation pathway for the syn
thymidine cyclobutane dimers. With a low collision energy, only loss one 2-deoxyribose was
observed. However, the abundance of the ions corresponding to the loss second sugar ring
were found increase with the collision energy. It should be mentioned that the ion of the
protonated 2-deoxyribose is observed for collision energy higher than 25 V. Interestingly,
the MS4 spectrum of the cis-syn cyclobutadithymidine acquired on the ion
trap spectrometer was identical to the MS2 spectrum of the corresponding
thymine photoproduct. This was expected since MS3 spectrometry yielded the
pseudo-molecular ion of the thymine photoproduct form the related thymidine derivative by
successive loss of the two 2-deoxyribose units. This example shows the interest of the ion
trap technology in the characterization of complex molecules.
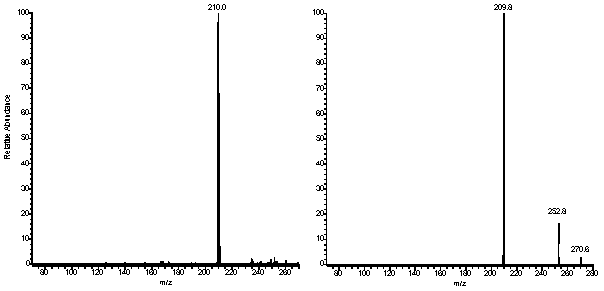
Figure 3: Ion trap mass spectra. left panel: MS2
mass spectrum of the thymine cis-syn cyclobutane dimer (parent ion: 253.1)
Right panel: MS4 mass spectrum of thymidine cis-syn cyclobutane dimer
(parent ion: 485.1-369.1-253.1). The difference in the relative abundance of the ions is
due to a different setting of the collision energy (13 and 11%, respectively).
As already observed with thymine photoproducts, the anti thymidine cyclobutane
dimers exhibited different spectra from the syn diastereoisomers (Table 2). Indeed,
a significant signal corresponding to protonated thymidine (m/z=243) was observed together
with the ion arising from the loss of the 2-deoxyribose units. Even at low collision
energy, two other ions were observed, corresponding to the protonated 2-deoxyribose
(m/z=117) and to protonated thymine (m/z=127), respectively. The latter ions is likely to
arise from the fragmentation of thymidine. Indeed, a complete loss of the 2-deoxyribose
ring is observed on the MS-MS spectrum of pure thymidine acquired under the conditions
used for the analysis of the photoproducts. These observations confirm that the
cyclobutane ring is more stable in the syn cyclobutadithymidine than in the anti
diastereoisomers. It should be pointed out that the six thymidine cyclobutane dimers have
already been extensively studied by FAB-mass spectrometry in both the positive and the
negative mode [19]. In contrast to the present results, no major difference was observed
between the mass spectra of the different diastereoisomers which exhibited protonated
thymine as the main fragmentation product. This can be explained by the higher energy
provided to the molecule by the FAB ionization technique, leading to a less selective
fragmentation. This comparison illustrates another advantage of the soft electrospray
ionization technique.
ion |
cis-syn |
(+) cis-anti |
(-) cis-anti |
(+) trans-syn |
(-) trans-syn |
trans-anti |
485 |
2 |
8 |
10 |
10 |
6 |
2 |
369 |
29 |
19 |
24 |
65 |
72 |
11 |
351 |
n.d. |
n.d. |
n.d. |
5 |
11 |
n.d. |
333 |
8 |
12 |
5 |
8 |
10 |
5 |
321 |
20 |
n.d. |
n.d. |
5 |
12 |
n.d. |
253 |
82 |
60 |
51 |
62 |
65 |
15 |
243 |
n.d. |
3 |
11 |
n.d. |
n.d. |
14 |
127 |
n.d. |
55 |
42 |
2 |
5 |
1 00 |
117 |
100 |
100 |
100 |
100 |
100 |
81 |
Table 2: Relative abundance of main ions
observed on the fragmentation spectrum of the diastereoisomers of the thymidine
cyclobutane dimers. The experimental conditions were identical for all dimers. In
particular, the argon pressure in the collision cell and the collision energy were set at
8x10-4 mTorr and 25 V, respectively.
3.3 Fragmentation of modified dinucleoside monophosphates and dinucleotides
The photoproducts of TpT and pTpT were also analyzed by ES/MS-MS in the positive mode. In addition, the presence of the easily ionized phosphate groups allowed their characterization as negatively charged ions. It should be mentioned that not all possible diastereoisomers could be isolated. Only one (6-4) photoproduct isomer is obtained upon exposure of TpT and pTpT to far-UV light. No anti cyclobutadipyrimidine diastereoisomers can be obtained because of the fixed parallel orientation of the C5-C6 double bond of the thymine rings. The cis-syn cyclobutane dimer is the major cyclobutane dimer obtained with both TpT and pTpT while one trans-syn isomer is produced is lower yield. The minor trans-syn II diastereoisomer of TpT described by Kao and co-workers [29] could not be isolated, likely because of too low amount of starting material.
A general trend is that the fragmentation spectra of TpT and pTpT photoproducts are
richer in the positive than in the negative mode. This was clearly illustrated by the
observation that, within the range of collision energy applied to the other photoproducts,
no fragmentation was observed for the cis-syn cyclobutane dimer of pTpT in the
negative mode. Use of higher collision energy led to a decrease in the signal of the
pseudo-molecular ion with no appearance of fragment signals. In contrast, peaks at m/z=431
and m/z=449 were observed in a relative abundance of 30% in the mass spectrum of the
latter photoproduct recorded in the positive mode (Fig. 4). Similarly, the
pseudo-molecular ion was the only signal observed on the MS-MS spectrum of the (6-4)
photoproduct of pTpT recorded in the negative mode with a collision energy of 15 V. The
latter value had to be raised to 30 V to induce significant fragmentation. In contrast,
dehydration was found to take place for a collision energy of 15 V in the positive mode.
These observations can be explained by the fact that positively charged pseudo-molecular
ions are obtained by protonation of either the base or the sugar moiety. This is likely to
destabilize the molecule and favor its fragmentation. In contrast, the negative ions arise
from the ionization of the phosphate group while the nucleosidic part remains unchanged.
It can also be added that TpT photoproducts have been analyzed by thermospray/mass
spectrometry in both the positive and the negative modes [21]. The thermospray spectra
exhibit a higher number of fragments with lower masses when compared to the ES/MS-MS data.
This is in agreement with the observation made with thymidine photoproducts that ES/MS-MS
is a soft ionization technique which allows a better control of the fragmentation of the
analyzed compounds.
Figure 4: Mass spectra of pTpT photoproducts
recorded on a triple quadrupole Quattro II spectrometer. The argon pressure in the
collision cell was set to 9x10-4 mTorr and the collision energy to 15 V for
the spectra recorded in the positive mode (upper and central panels) and 30 V for the
spectrum recorded in the negative mode (lower panel).
The mass spectra of the (6-4) photoproducts of TpT and pTpT recorded in the positive mode exhibit mainly fragments arising from the dehydration of the 5'-end thymine, as observed for the related base lesion. This indicates that the presence of a phosphodiester bond in modified TpT and pTpT reinforces the N-glycosidic bond. Indeed, loss of the 2-deoxyribose unit was the main fragmentation observed with thymidine photoproducts (vide supra). The fragmentation spectrum of the Dewar valence isomer also exhibits the dehydrated ion as the main products, together with several unidentified peaks at m/z<250. The interpretation of the fragmentation of the cyclobutane dimers of TpT and pTpT is less obvious. Indeed, the main ions observed upon fragmentation of the [M+H]+ pseudo-molecular ion of TpT cis-syn and trans-syn cyclobutane dimers correspond to the loss of two 98 and two 116 uma. This leads to the formation of ions at m/z= 449, 431, 351 and 333 uma. A first explanation is the loss of fragments of the 2-deoxyribose units. Indeed, 98 uma corresponds to the mass of a 2-deoxribose ring which has lost an hydroxyl group involved in the phosphodiester bond. An alternative fragmentation pathway may involve the loss of CO-NH-CO-C-CH3 fragments from the pyrimidine ring on both the 5' and 3' end. Loss of molecule of water from the [MH-98]+ ion could account for the [MH-116]+ ion. Support for the latter reaction was provided by the MS3 analysis of the cis-syn of the trans-syn cyclobutane dimers on the ion trap spectrometer. Indeed, fragmentation of the ion at m/z=449 gives rise to the ion at m/z= 431. The ions at m/z= 449 and 431 are the also the main peaks observed on the spectrum of the pTpT cis-syn cyclobutane dimer. Again, this could be explained by either the loss of a phosphorylated 2-deoxyribose ring or of a phosphate group and a CO-NH-CO-C-CH3 fragment. Definitive conclusions of the fragmentation pattern could only be achieved by the analysis of isotopically labeled photoproducts.
The interpretation of the spectra recorded in the negative mode was facilitated by the
reduced number of product ions (Fig. 5). The main signal on the spectrum of the (6-4)
photoproducts of TpT and pTpT and their Dewar valence isomers was at [M-H-113]-.
This can be accounted for by a fragmentation of the 5' saturated pyrimidine ring.
Interestingly, no dehydration reaction was observed in contrast to the results obtained in
the positive mode. As already mentioned, no significant fragmentation could be obtained
for the cis-syn cyclobutane dimer of pTpT. In contrast, the corresponding TpT
photoproduct yielded a predominant ion at m/z=447 corresponding to the loss of a 98 uma
fragment as observed in the positive mode. The same rearrangement was also observed for
the trans-syn isomer. It can be added that a significant ion at m/z= 195 was
observed for all TpT derivatives, which corresponds to a 2-deoxyribose monophosphate
moiety.
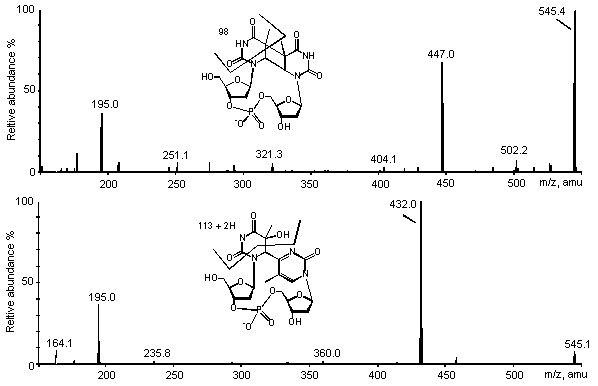
Figure 5: ES/MS-MS spectra of the cis-syn
cyclobutane dimer (upper panel) and the (6-4) photoproduct (lower panel) of TpT. Spectra
were recorded in the negative mode on a triple quadrupolar API 3000 spectrometer.
Detection conditions were identical for both analyses.
3.4 Detection of TpT photoproducts by HPLC coupled to tandem mass spectrometry
Even though the cis-syn cyclobutadithymine was identified almost 40 years ago, one still lacks a method for the simultaneous detection of the main base photolesions within DNA. The formation of cyclobutadithymine has been monitored by using formic acid hydrolysis of [3H]-thymine labeled DNA followed by a chromatographic separation [30-32]. Cis-syn cyclobutane dimers have also been measured within acid hydrolyzed DNA by applying a GC-MS isotopic dilution technique [33]. The direct detection of (6-4) photoproducts is hampered by their low stability under usual acidic hydrolysis conditions. However, the rate of formation of the latter lesions was determined within DNA using a mild HF/pyridine hydrolysis followed by a HPLC/fluorescence detection [34]. However, none of these techniques allows the simultaneous measurement of the cyclobutane dimers, the (6-4) adducts and the Dewar valence isomer. The bulk of the results obtained on the formation of far-UV induced dimeric photoproducts within isolated and cellular DNA has been obtained by using indirect techniques including immunological approaches [35-37] and use of repair enzyme as a way to convert the lesions into easily detectable DNA strand breaks [38]. Even though they are highly sensitive, the latter methods do not allow the individual quantification of each bipyrimidine lesion for a given class of photoproducts. Therefore, a technique allowing the simultaneous detection of the three types of photoproducts arising from the different bipyrimidine sequences is still needed.
The results presented above show that the use of an electrospray ionization technique
is particularly suitable for the mass spectrometry analysis of the various thymine
photoproducts. Therefore, the latter technique combined to liquid chromatography might
provide a new and efficient analytical tool for the detection of far-UV induced thymidine
photoproducts in hydrolyzed DNA. Interestingly, TpT photoproducts are well separated by
reverse-phase HPLC. In addition, the latter compounds can be analyzed in the negative mode
which is more sensitive than the detection of positive ions because of a reduced
background. Therefore, a method was optimized for the analysis of TpT photoproducts by
HPLC coupled to a triple quadrupolar spectrometer used in the negative mode with a MRM
detection. A 2 mm column was used to avoid splitting of the HPLC eluent and a gradient was
applied to provide an optimal separation. As already mentioned, the major signal of the
fragmentation spectrum of the TpT cyclobutane dimers was observed at m/z=447 in the
negative mode. In contrast, the pseudo-molecular ion of the (6-4) photoproduct and of its
Dewar valence isomer yielded a daughter ion at m/z=432. The spectrometer was thus set to
monitor the transitions 545-447 and 545-432. This analytical technique allowed the
simultaneous detection of a mixture of the four photoproducts (Fig. 6). Indeed, the (6-4)
adduct and the trans-syn cyclobutane dimer were separated as single peaks with a
high proportion of methanol in the mobile phase. In contrast, the Dewar valence isomer and
the cis-syn cyclobutane dimer were eluted close to each other. However, their
difference in favored fragmentation pathway allowed to resolve them anyway. The
sensitivity of the detection is high since a signal to noise ratio of 10 is obtained for
200 fmol of product injected on the column. The detection of TpT photoproducts thus
appears to be an interesting approach for the measurement of far-UV induced DNA lesions.
The key point remains the optimization of the release of the photoproducts from DNA as
dinucleoside monophosphates. This might be achieved by the use of both 3' and 5'
phosphodiesterases. Indeed, cyclobutane dimers and (6-4) photoproducts were found to be
quantitatively released as dinucleoside monophosphate from far-UV irradiated short
oligonucleotides [23,39].
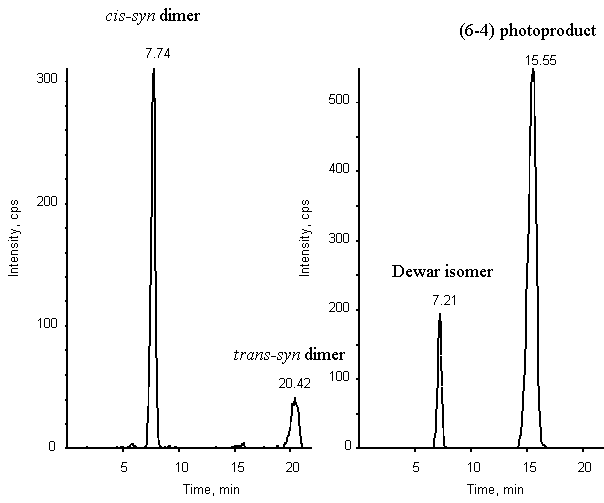
Figure 6: Chromatogram of a mixture of the cis-syn
and trans-syn cyclobutane dimers, (6-4) photoproduct and Dewar valence isomer of
TpT (2 pmol of each compound). Detection was provided by a triple quadrupolar spectrometer
API 3000 used in the MRM mode. Two transitions were monitored: 545-447 (left panel) and
545-432 (right panel).
Conclusion
The present study allows two interesting comparisons. First, it clearly shows the
advantages of the ES/MS-MS approach for the acquisition of fragmentation spectra of
thymine photoproducts. Indeed, when compared with previous data based on the use of FAB or
thermospray ionization, the ES/-MS-MS spectra show a more controlled fragmentation. No
differences in fragmentation pattern was observed between the (6-4) adducts and their
Dewar valence isomers. However, a significant influence of the diastereoisomery of the
cyclobutane dimer was observed for both the thymine and the thymidine derivatives. In
addition, a comparison could be done between the triple quadrupole and the ion trap
technology. The latter approach was unambiguously more efficient for the determination of
the fragmentation pathways. It also appeared to be slightly more sensitive for the
recording of full spectra than the triple quadrupolar spectrometers. However, the latter
system provided a very sensitive detection when used in the MRM mode which, in contrast,
only slightly improved the sensitivity of the ion trap spectrometer.
Acknowledgment
The authors wish to thank Colette Lebrun (DRFMC/SCIB, Grenoble); Claude Bosso (CERMAV, Grenoble) and Bernard Brasme (CRSSA, La Tronche) for fruitful discussions on the optimization of the Quattro II spectrometer.
References
[1] H.S. Black, F.R. de Gruijl, P.D. Forbes, J.E. Cleaver, H.N. Ananthaswany, E.C. de Fabo, S.E. Ullrich, R.M. Tyrrell, Photocarcinogenensis: an overview, J. Photochem. Photobiol. B: Biol. 40 (1997) 29-47.
[2] J. Cadet, P. Vigny. (1990) In Morrison, H. (ed.), Bioorganic Photochemistry. Wiley, New York, Vol. 1, pp. 1-272.
[3] T. Douki, J. Cadet. (1995) In Jornvall, H., and Jollčs, P. (eds.), Interface between chemistry and biochemistry. Birkhauser Verlag AG, Basel, pp. 173-197.
[4] J.E. Donnellan, R.B. Setlow, Thymine photoproducts but not thymine dimers found in ultraviolet-irradiated bacterial spores, Science 149 (1965) 308-310.
[5] R. Beukers, W. Berends, Isolation and identification of the irradiation product of thymine, Biochim. Biophys. Acta 41 (1960) 550-551.
[6] D. Weinblum, H.E. Johns, Isolation and properties of isomeric thymine dimers, Biochim. Biophys. Acta 114 (1966) 450-459.
[7] A.J. Varghese, S.Y. Wang, Thymine-thymine adduct as a photoproduct of thymine, Nature 160 (1968) 186-187.
[8] G.J. Fisher, H.E. Johns, Ultraviolet photochemistry of thymine in aqueous solution, Photochem. Photobiol. 11 (1970) 429-444.
[9] A.J. Varghese, Photochemistry of thymidine on ice, Biochemistry 9 (1970) 4781-4787.
[10] W.A. Franklin, P.W. Doetsch, W.A. Haseltine, Structural determination of the ultraviolet light-induced thymine-cytosine pyrimidine-pyrimidone (6-4) photoproduct, Nucleic Acids Res. 13 (1985) 5317-5325.
[11] R.E. Rycyna, J.L. Alderfer, UV irradiation of nucleic acids: formation , purification and solution conformational analysis of the '6-4' lesion of dTpdT, Nucleic Acids Res. 13 (1985) 5949-5963.
[12] J.-S. Taylor, M.P. Cohrs, DNA, light, and Dewar pyrimidones: the structure and biological significance of TpT3, J. Am. Chem. Soc. 109 (1987) 2834-2835.
[13] L.-S. Kan, L. Voituriez, J. Cadet, Nuclear magnetic resonance studies of cis-syn, trans-syn, and 6-4 photodimers of thymidylyl(3'-5')thymidine and cis-syn photodimers of thymidylyl(3'-5')thymidine cyanoethyl phosphotriester, Biochemistry 27 (1988) 5796-5803.
[14] J.-S. Taylor, H.-L. Lu, J.J. Kotyk, Quantitative conversion of the (6-4) photoproduct of TpdC to its Dewar valence isomer upon exposure to simulated sunlight, Photochem. Photobiol. 51 (1990) 161-167.
[15] T. Douki, L. Voituriez, J. Cadet, Characterization of the (6-4) photoproduct of 2'-deoxycytidylyl-(3'-5')-thymidine and of its Dewar valence isomer, Photochem. Photobiol. 27 (1991) 293-297.
[16] T. Douki, J. Cadet, Far-UV photochemistry and photosensitization of 2'-deoxycytidylyl-(3'-5')-thymidine: isolation and characterization of the main photoproducts, J. Photochem. Photobiol. B: Biol. 15 (1992) 199-213.
[17] T. Douki, J. Cadet, Formation of cyclobutane dimers and (6-4) photoproducts upon far-UV photolysis of 5-methylcytosine containing dinucleoside monophosphates, Biochemistry 33 (1994) 11942-11950.
[18] J. Ulrich, R. Teoule, R. Massot, A. Cornu, Etude de la fragmentation de dérivés de l'uracile et de la thymine par spectrométrie de masse, Org. Mass Spectrom. 2 (1969) 1183-1199.
[19] J. Cadet, L. Voituriez, F.E. Hruska, L.-S. Kan, F.A.A. de Leeuw, C. Altona, Characterization of thymidine ultraviolet photoproducts. Cyclobutane dimers and 5,6-dihydrothymines, Can. J. Chem. 63 (1985) 2861-2868.
[20] R.L. Hettich, M.V. Buchanan, C.-H. Ho, Characterization of photo-induced pyrimidine cyclobutane dimers by laser desorption Fourier transform mass spectrometry, Biomed. Environ. Mass Spectrom. 19 (1990)
[21] R. Bérubé, D.G.E. Lemaire, B.P. Ruzsicska, Thermospray high-performance liquid chromatography analyses of the photoproducts of dTpdT and dTpdU, Biol. Mass Spectrom. 21 (1992) 259-266.
[22] J.-L. Ravanat, T. Douki, M.-F. Incardona, J. Cadet, HPLC separations of normal and modified nucleobases and nucleosides on an amino silica gel column, J. Liq. Chromatog. 16 (1993) 3185-3202.
[23] T. Douki, T. Zalizniak, J. Cadet, Far-UV-induced dimeric photoproducts in short oligonucleotides: Sequence effects, Photochem. Photobiol. 66 (1997) 171-179.
[24] C.L. Greenstock, H.E. Johns, Photosensitized dimerization of pyrimidines, Biochem. Biophys. Res. Com. 30 (1968) 21-27.
[25] C. Hélčne, M. Charlier, Photosensitized reactions in nucleic acids. Photosensitized formation and splitting of pyrimidine dimers, Biochimie 53 (1971) 1175-1180.
[26] J. Blais, T. Douki, P. Vigny, J. Cadet, Fluorescence quantum yield determination of pyrimidine (6-4) pyrimidone photoadducts, Photochem. Photobiol. 59 (1994) 404-404.
[27] M. Charlier, C. Hélčne, Photochemical reactions of aromatic ketones with nucleic acids and their components-III. Chain breakage and thymine dimerization in benzophenone photosensitized DNA, Photochem. Photobiol. 51 (1972) 527-536.
[28] R.O. Rahn, J.L. Hosszu, Influence of relative humidity on the photochemistry of DNA films, Biochim. Biophys. Acta 190 (1969) 126-131.
[29] J.L.-F. Kao, S. Nadji, J.-S. Taylor, Identification and structure determination of a third cyclobutane photodimer of thymidylyl-(3'-5')-thymidine: the trans-syn II product, Chem. Res. Toxicol. 6 (1993) 561-567.
[30] M.H. Patrick, Studies on thymine-derived UV photoproducts in DNA-I. Formation and biological role of pyrimidine adducts in DNA, Photochem. Photobiol. 25 (1977) 357-372.
[31] J. Cadet, N.E. Gentrer, B. Rozga, M.C. Paterson, Rapid quantification of ultraviolet induced thymine-containing dimers in human cell DNA by reverse-phase high performance liquid chromatography, J. Chromatogr. 280 (1983) 99-108.
[32] H.J. Niggli, P.A. Cerutti, Cyclobutane-type pyrimidine dimer formation and excision in human skin fibroblasts after irradiation with 313 nm ultraviolet light, Biochemistry 22 (1983) 1390-1395.
[33] I.D. Podmore, M.S. Cooke, K.E. Herbert, J. Lunec, Quantitative determination of cyclobutane dimers in DNA by stable isotope-dilution mass spectrometry, Photochem. Photobiol. 64 (1996) 310-315.
[34] T. Douki, L. Voituriez, J. Cadet, Measurement of pyrimidine (6-4) pyrimidone photoproducts in DNA by a mild acidic hydrolysis-HPLC fluorescence assay, Chem. Res. Toxicol. 8 (1995) 244-253.
[35] D.L. Mitchell, B.S. Rosenstein, The use of specific radioimmunoassays to determine action spectra for the photolysis of (6-4) photoproducts, Photochem. Photobiol. 45 (1987) 781-786.
[36] T. Mori, T. Matsunaga, C.-C. Chang, J.E. Trosko, O. Nikaido, In situ (6-4) photoproduct determination by laser cytometry and autoradiography, Mutat. Res. 236 (1990) 99-105.
[37] W.M. Olsen, H.S. Huitsfeldt, G. Eggset, UVB-induced (6-4) photoproducts in hairless mouse epidermis studied by quantitative immunohistochemistry, Carcinogenesis 10 (1989) 1669-1673.
[38] D.C. Thomas, D.S. Okumoto, A. Dancar, V.A. Bohr, Preferential DNA repair of (6-4) photoproducts in the dihydrofolate reductase gene of Chinese hamster ovary cells, J. Biol. Chem. 264 (1989) 1374-1378.
[39] M. Liuzzi, M. Weinfeld, M.C. Paterson, Enzymatic analysis of isomeric
trithymidilates containing ultraviolet light-induced cyclobutane pyrimidine dimers I
Nuclease P1 mediated hydrolysis of the intradimer phosphodiester linkage, J. Biol. Chem.
264 (1989) 6355-6363.