| Papers and Posters | Site Home Page |
The photochemical behaviour of 6-X-4H-3-bicyclo[2.2.1]-5-heptene-
-2,3-dicarboimidoiminomethyl)-4-chromones and -4-chromenone.
Photochromism and Thermochromism
Anton Gáplovskýa, Jana Donovalováa, Margita Lacováb and Renáta Mracnováa Jarmila Chovancováb
aInstitute of Chemistry Faculty of Natural Science Comenius University, Mlynska dolina CH2, 842 15 Bratislava, Slovakia
b
Department of Organic Chemistry Faculty of Natural Science Comenius University, Mlynska dolina CH2, 842 15 Bratislava, SlovakiaaInstitute of Chemistry Faculty of Natural Science Comenius University, Mlynska dolina CH2, 842 15 Bratislava, Slovakia
b
Department of Organic Chemistry Faculty of Natural Science Comenius University, Mlynska dolina CH2, 842 15 Bratislava, Slovakia
Abstract
The photochemical behaviour of 6-X-4H-3-bicyclo[2.2.1]-5-heptene-2,3-dicarboimidoiminomethyl)-4-chromones (X = CH3, Cl, NO2 ) and -4-chromenone in CH3OH, benzene upon irradiation (l irr = 310 nm) was studied. Direct E-Z or Z-E photoisomerization and thermal Z-E isomerization were observed. The activation energies of Z-E isomerization were in the range 8.7–69.5 kJ.mol-1. Fluorescence emission and excitation spectra were measured. Prolongation of the irradiation of the solution of the title copmpounds. After the equilibrium between E and Z isomers is reached, resulting in the irreversible process. The products of this irreversible process were identified.
1. Introduction
Compounds possessing the exocyclic C=N bond as well as the C=C and N=N bonds respectively undergo the isomerization. The conversion from one isomer to the other can be initialized by light or by temperature [1]. In the case that the E-Z isomerization is reversible photochemical process and both isomers have different absorption spectra, this phenomenon is called photochromism [2].
Besides E-Z isomerization about the C=N double bond, the possibility of conformer and tautomer formation should be considered in dependence on the structure of fragments. Structure of the fragment adjacent to C=N double bond can effect photochemical or thermal reactivity of compounds with photochromic properties. In this paper we describe the photochemical system where photochromism depends on the geometric isomerization of substituted 6-X-4H-3-bicyclo[2.2.1]-5-heptene-2,3-dicarboimido-iminomethyl)-4-chromones (Fig. 1.). The photochemical reactivity of these compounds and thermochromism were studied.

(I) X = Cl; (IV)
(II) X = CH3;
(III) X = NO2;
Fig. 1. Structure of studied compounds.
2. Experimental details
2.1. General methods
All photolysis experiments were carried in quartz spectrophotometric cell using the filtered irradiation (l = 310 nm) from an UVP, Inc. Chromato VUE transilluminator, Model TM-15 or unfiltered light using from an Osram halogen lamp (150 W) in degassed methanol or benzene under nitrogen at room temperature. The course of photolysis was monitored by UV, VIS spectrophotometer HP 8452A Array spectrophotometer. After irradiation, the reaction mixture was concentrated under vacuum and analysed by gas chromatography. GC analysis of the samples was performed on a Helwett-Packard (HP) (Palo Alto, CA, USA) 5890A Series II gas chromatograph equipped with split-splitless injector (300 oC, spliting ratio 1:30, splitless time 1’). Helium (TATRAGAS, 99.95%) was used as the carier gas (inlet pressure 50 kPa). The injection volume was 1 m l. An HP 5890A Series II gas chromatograph interfaced to an HP 5917A mass-selective detector with an HP MS Chemstation data system was used for identification of the GC components. The column used was a cross-linked fused-silica capilary column (10m ´ 0.32mm I.D.) coated with polydimethylsiloxane (0.20 m m phase thickness). The oven temperature was programed from 40 oC to 320 oC at 15 oC min-1. The temperatures of the isolation chamber and of the transfer line were 180 oC and 280 oC, respectively. The electron energy was 70 eV. Mass spectra and reconstructed total ion chromatograms were obtained by automatic scanning in the mass range m/z 45-450 at 2.2 scans s-1. 1H-NMR spectra and 13C NMR spectra were recorded in DMSO with VARIAN GEMINI 2000, 300 MHz NMR spectrometer, using tetramethylsilane (TMS) as internal standard. UV spectra were recorded with HP 8452A Array spectrophotometer. Elemental analysis were performed with Carlo-Erba CHN 1106.
2.2. Materials
Methanol and benzene (spectroscopic grade, Merck) were used as solvents. The starting compunds 6-X-4H-3-bicyclo[2.2.1]-5-heptene-2,3-dicarboximidoimino-methyl-4-chromones and -4-chromenes were synthetized according the following procedure: Solution of N-aminobicyclodicarboximide (0.01 mol) in small amount of ethanol was added to ethanol solution of 3-formylchromones (0.01 mol) and catalytic amount of p-toluenesulfonic acid. The mixture was stirred at the room temp. for 2 h. The solid compound was separated, washed with diethylether and recrystallized from xylene. NMR spectra, elementar analysis and melting points were utilized to established the purity of the compounds.
3. Results and discussion
The studied compounds absorb UV light in the range 220 - 430 nm. With increasing polarity of solvent the absorption maximum is red shifted by a few nm. This absorption corresponds to p ,p * band which overlap n,p * band. Electron-donor Substituent effects on the position of long-wavelength absorption is given in Table 1.
TABLE 1
Basic characteristics of the absorption and fluorescence spectra of I, II, III and IV in methanol
Compound |
Absorbance l max [nm] log e [l mol cm-1] |
Fluorescence l max [nm] |
||
I |
348* 311 261 |
3.76 4.13 4.18 |
435 |
|
II |
308 260 |
4.15 4.28 |
424 |
|
III |
330 314 |
4.19 4.19 |
421 |
|
IV |
326* 292 226 |
4.07 4.37 4.36 |
436* 398 |
|
* shoulder
Substitution of phthalimide for NH-Ph fragment in the molecule has much more influence on absorption spectra (l max = 355 nm in CH3OH). This change of absorption spectra can be caused by the different electron-donor ability of both fragments. The fluorescence emission of the studied compounds has a very low intensity in the range of l = 350-550 nm (Fig. 2). Maximum of the fluorescence emission is red shifted with electron-donor ability of the substituent. Intensity of fluorescence emission in benzene is lower compare to methanol and increases with decreasing temperature. Maximum is blue shifted (~ 4 nm). We were interested also to find out which part of the structure I-IV is important for the observed fluorescence. Spectra of parent chromone aldehydes were measured therefore. The fluorescence of the corresponding chromone aldehydes compare to the fluorescence of I, II, III and IV has nearly the same position and shape (Fig. 3.) but they are different only in the intensity of fluorescence. For example, the intensity of the fluorescence of 6-methylchromone-3-carboxaldehyde is 20 times higher than intensity of the fluorescence of II. From the mentioned above follows that the fluorescence occurs from the singlet state which is localised on the chromone fragment of II The fluorescence spectrum of N-aminobicyclo[2.2.1]-5-heptenedicar-boximide) is blue shifted ~ 60 nm compare to II or 6-methylchromone-3-carboxalde-hyde, respectively (Fig. 3.).

Fig. 2. Fluorescence emission and absorption spectra of the E-isomers of I (red), II (blue), III (black) and IV (green) in methanol (l ex = 310 nm)
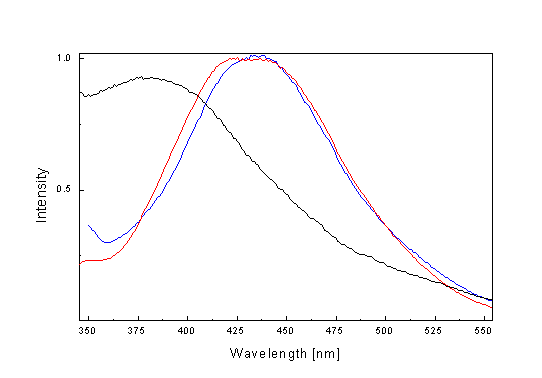
Fig. 3. Fluorescence emission spectra (l ex = 310 nm) of II ( blue) correspond 6-methylchromone-3-carboxaldehyde (red) and N-aminobicyclo[2.2.1]-5-heptenedicarboximide (black).
Excitation spectra of the chromones depend on the wavelength of the emission (Fig. 4.). Changes in excitation spectra are caused by the photolability of the E-isomer

Fig. 4. Fluorescence excitation spectrum of the E-isomer of I in methanol [l em = 420 nm (black), 450 nm (red), 480 nm (blue), 540 nm (green)].
as well as the presence of their conformers in the solution which can be
stabilized by intramolecular interactions as the interaction of H in position 2 of
chromone fragment or H of HC=N with C=O of chromone or phthalimide fragments,
respectively. UV irradiation of the studied compounds at l =
310 nm in CH3OH or benzene at the room temp. causes the blue shift of the
absorption maximum as well as decreases of

Scheme 1
the absorption of long-wavelength band (Fig. 5.). The change of long-wavelength absorption corresponds to E-Z geometric isomerization about the C=N double bond (Scheme 1). This isomerization is a reversible process. The back Z-E isomerization occurs photochemically with the visible light in the polar (e.g. CH3OH) or in the non polar solvent (e.g. benzene) and thermally only in polar solvent. The molecular structure or substituent has a very small effect on the photochemical isomerization compare to thermal, where the effect is substantial.

Fig. 5. UV spectrum of the E-isomer of II in methanol (c = 10-4 mol.dm-3) at the different time of irradiation. (0 s - orange, 5 s - red, 10 s - green, 600 s - black).
We have also calculated the activation energies of the studied isomerization [Tab. 2.]. As can be seen from Table 2. the activation entropy is enhanced in the same direction as the activation energy. The negative activation entropies show, that the transition state is less organized than initial state [3] The change of the activation entropy can be
TABLE 2.
Termodynamic data of the Z-E isomers of I, II, III and IV in methanol
Compound |
D E¹ [kJ.mol-1] |
D S¹ [J.mol-1K-1] |
I |
40,4 |
-133,2 |
II |
50,9 |
-105,5 |
III |
8,7 |
-236.4 |
IV |
69,5 |
-54,5 |
explained by steric hindrance of the substituent. The major contribution has internal rotation entropy, which characterize intramolecular rotation about the bonds in molecule.
The solvatation ability and polarity of the solvent play important role in the thermal isomerization. In contradiction to isomerization in CH3OH, thermal isome-rization was not observed in the range 19-58 oC. The long-wavelength absorption in UV spectra in benzene is blue shifted by ~ 4 nm (l max = 310 nm). From the small shift in absorption spectra follows that the change of energy is small and can hardly block thermal isomerization. A rapid isomerization was detected in CH3OH but a very slow one in hexane. Optimalization of the geometry of E and Z isomers by Hyperchem 3 confirm the assumption that these isomers are not planar. E and Z isomers have chromone fragment and C=N bond in one plane and phthalimide fragment is out of this plane. The lone pair of nitrogen of the Z-isomer can interact with p-electrones of phthalimide carbonyl group [4]. Substituent effects the electrone density of the lone pair of the nitrogen atom as well as the strength of interaction. The strength of this interaction has influence on the thermal stability of the Z isomer. Light of the halogen lamp which is absorbed by the Z-isomer has enough energy to initialize Z-E isome-rization in benzene.
The change of absorption of the E-isomer occurs very fast upon UV irradiation. After ~ 15 s irradiation, the equilibrium of the E and Z isomers is reached in both solvent. Applaing the light of the halogen lamp on the reaction mixture, Z-E isomerization occurs and the absorption returns to the initial state. This process was repeated many times (Fig. 6.). Prolongation of irradiation (l = 310 nm, t = 50 min.)

Fig. 6. Change of UV absorbance (l = 308 nm) of II after alternate irradiation by UV light (310 nm, D = 15 s) and VIS light (halogen lamp, D = 30 s).
has a small effect on the UV spectra. Z-E isomerization upon VIS light does not occur at this stage of the reaction. Intensity of UV absorbance as well as intensity of fluorescence decrease during the photochemical E-Z isomerization. Intensity of the fluorescence enhances with prolongation of the irradiation contrary to the intensity of UV absorbtion. Time behaviour of emission and excitation spectra at the different l is showed on Fig. 7. From the spectral changes follow that after the E-Z equilibrium is reached the other photochemical changes occur. They are irreversible. We can not decide if E or Z isomer undergoes to the additional photochemical conversion. The proof can not be reached by separation methods as GC, HPLC either because the equilibrium process between E-Z isomers is so fast to compare with competitive photochemical reactions. If we consider that chromone fragment of the E and Z isomers and C=N group are in the same plane then the hydrogen on CH=N bond and

Fig. 7. Change of absorbance UV ( l = 308 nm, blue) and excitation (l = 350 nm, red; l = 300 nm, green) spectra of II in methanol in dependence of the time of irradiation.
the hydrogen in position 2 of chromone fragment are in the position when they can interact with the C=O of chromone or with the nitrogen of C=N group, respectively.
These interactions can be considered in the ground state too. The similar interaction

Fig. 8. Structure of products after photolysios of I - IV in methanol.
was described in salicylidene-2-aminopyridines [5]. Abstraction of hydrogen from the carbon of CH=N group by the C=O of phthalimide fragment is possible from photochemical point of view. This hydrogen is in g -position with respect to the C=O group. After the hydrogen abstraction a -cleavage occurs. In the reaction mixture were identified by GC chromatography following products V - XII (Fig. 8).
Literature
1 H. Gorner and E. Fisher; J. Photochem. Photobiol. A Chem., 57 (1991) 235
K. Appenroth, M. Reincherbächer and R. Paetzold; Z. Chem., 23 (1983) 149
J. Kessler, Angew. Chem., 82 (1970) 237
J. Kessler, Tetrahedron, 30 (1974) 1861
J. Elguero, R. Jacquier and C. Marzin; Bull. Soc. Chem. Fr., 1374 (1969)
E. Lambi, D. Gegiou and H. Hadjoudis; J. Photochem. Photobiol. A. Chem., 86 (1995) 241,
2 H. Durr and H. Bouas-Laurent, Photochromism, Elsevier, 1990.
3 R.W. Haywood in "Equilibrium Thermodynamics for Engineers and Scientists" John Wiley and Sons New York, Ltd. Chap., 12, 1980
4 M. R .Bell and S. Archer , J. Am. Chem. Soc., 82 (1960) 151
5 E. Hadjoudis, M. Vitorakis and I. Moustakali-Mavridis, Mol. Cryst. Liq. Cryst., 137 (1986) 1