| Papers and Posters | Site Home Page |
Two different fluorescent states of singly protonated dimeric forms
of tetraphenylporphine
Alexander V. Udal’tsov* and Yuri V. Kovalev
Faculty of Biology, Moscow State University, Leninskie Gory 119899 Moscow, Russia
Properties of ground and excited states of singly and doubly protonated dimeric forms of meso-tetraphenylporphine (TPP) in water-organic solutions are investigated by absorption and luminescence spectroscopy. The data indicating the existence of three protonated dimeric forms of TPP, doubly protonated porphyrin dimer with the maximum of Soret band at 437 nm and two different singly protonated dimers with lmax = 403 and 465 nm of Soret bands, are presented. The bands of these three dimeric forms in the red region of the absorption spectra are strongly overlapped that provides a base for interaction between them in the excited state. But only two different fluorescent states of the TPP dimers appears to be revealed with selective excitation of the dimeric forms of porphyrin (lex = 403, 437 or 465 nm). The corresponding maxima of the emission bands are observed at 693 and 730 nm in the fluorescence spectra. The results are interpreted in the terms of specific interactions of dimeric forms of porphyrin with water.
Key words: dimers, porphyrins, fluorescence properties, different fluorescent states, influence of solvent.
Introduction
Numerous model systems have been developed for mimicking of photosynthetic events taking place in living organisms with the aim of understanding of physico-chemical mechanisms of light energy conversion and storage as a fuel [1-4]. At the primary steps of photosynthesis one from the main role in the reaction center belongs to porphyrin pigments unique properties of which are studied extensively for many years [5-7]. Although properties of porphyrin pigments are well studied but in this paper we will concern some aspects of the properties of porphyrins that is still remained without an attention of researchers. As known for many years ago, cation radicals of porphyrins can produce isoporphyrins, the porphyrin tautomers, in the result of reaction with methanol [8]. Meanwhile on the other hand, there is another type of porphyrin tautomerization, NH- tautomerization, that was observed both in metal-free porphyrins and in covalently linked porphyrins too [9]. Isoporphyrins can be obtained as for metal-free porphyrin as well as for metalloporphyrin and moreover their properties [8, 10] appears to be some similar to singly protonated metal-free porphyrin dimers [11], where NH-tautomerization can play one from the crucial role because of water molecules are involved by these dimers in their dimeric complex.
Hence, these data suggest that interaction of a solvent with porphyrin has the specificity when the competition between the pathway of isoporphyrin formation and realization of NH-tautomerization can be occurred. On the other hand, there are data indicating water influence, namely, effects of the hydrogen network on the rate constants of energy migration, electron transfer and charge recombination in biological reaction center [12]. The data suggest an important role of protons relaxation of water-protein structure when the proton relaxation can influence on the state of porphyrin pigments interacting with their surrounding. In connection with the above, it was interesting to investigate the properties of different dimeric forms of porphyrin in the ground and in the excited states and to elucidate whether interaction between the different dimeric forms takes place, and whether a dependence there is from different conditions, i.e. the compose of water-organic solution, in particular, when an alcohol is utilized as the organic component.
Thus, in the present work ground and excited states of the dimeric forms of meso-tetraphenylporphine prepared in water-organic solutions are investigated by absorption and luminescence spectroscopy.
Experimental
Synthesis of meso-tetraphenylporphine (TPP) was carried out according to the procedures described elsewhere [13]. Tetrahydrofuran, dioxane and other organic solvents were additionally purified by conventional methods [14]. For the preparation of aqueous-organic solutions distilled or twice distilled water was used.
UV-visible spectra of porphyrin solutions were recorded with a Specord M-40 spectrophotometer. Fluorescence spectra were obtained with the setup described elsewhere [15]. The measurements were carried out under the same conditions as earlier [16] except for the excitation. The excitation in the region of Soret band was provided by monochromatic light with a half-width of 12 nm separated from xenon lamp with the use of monochromator. Fluorescence spectra were completely calibrated using the known spectral sensitivity of the equipment. All measurements were carried out at 298K.
Results
Figure 1 shows absorption spectra of monomeric TPP (curve 1) and TPP in aqueous-dioxane solution in the presence of 0.2 N HCl (curve 2), the state of this porphyrin was assigned to doubly protonated dimer [17]. The spectrum (curve 2) exhibits Soret band with the maximum at 437-438 nm and the band in the red region with the maximum at 654 nm. Note that this Soret band is some wider as compared with that in the spectrum (curve 1). It was found that about 10% and higher contents of water in organic solvent brings about dimerization of porphyrin and a high value of dimerization constant was calculated for equilibrium conditions [17]. On the other hand, doubly protonated state of the dimer was suggested on the base of the following results. Initially we tried to find singly protonated monomeric porphyrin. It was proposed that interaction between charged porphyrin molecules can be excluded under porphyrin bounding with polymer, when the ratio of monomer chain to the bound porphyrin molecule is about of three order of magnitude.
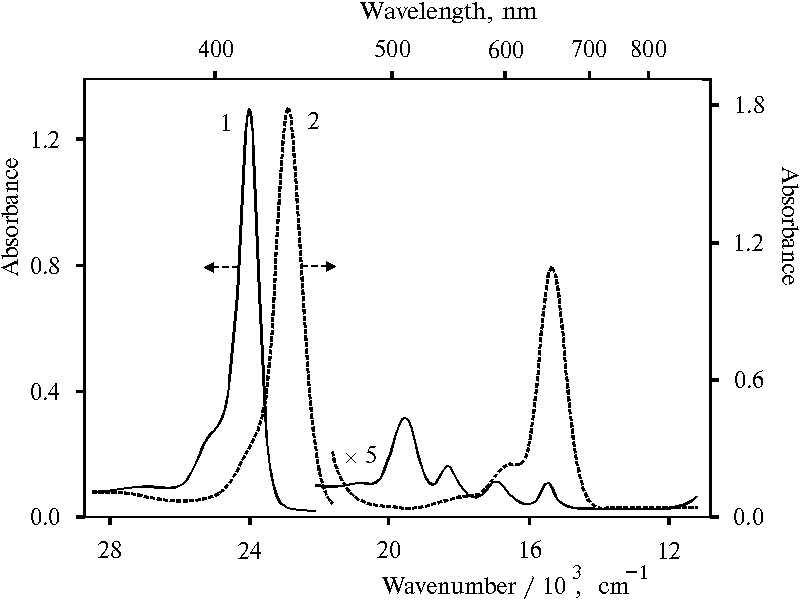
Fig. 1. Absorption spectra of meso-tetraphenylporphine in dioxane, 1; and in 50% aqueous solution of dioxane (v/v) in the presence of 0.2 N hydrochloric acid, 2.
The absorption spectra of porphyrin-polymer sample with similar ratio (see scheme 1 in [18]) are presented in Fig. 2. According to the previous data, more than 90% of the bound porphyrin is present in monomeric state in this sample [18]. The spectrum of porphyrin bound with polymer in water (curve 1) exhibits two absorption bands with the maxima at 445 and 654 nm. Therefore, this porphyrin at about 3 pH is fully, i.e. doubly, protonated in water. There is a small bend in titration curve of this sample at about 5.5 pH [19]. This small bend appears to mean the removing of one proton from the doubly protonated monomeric porphyrin under titration with alkaline solution. Hence, the next large bend in the titration curve have to characterize transition of singly protonated porphyrin into neutral its molecule. An intermediate spectrum containing about half of porphyrin concentration in the neutral form is presented in Fig. 2, curve 2. This spectrum exhibits Soret band of protonated porphyrin with the maximum at 439 nm and red band with the maximum at 649 nm.
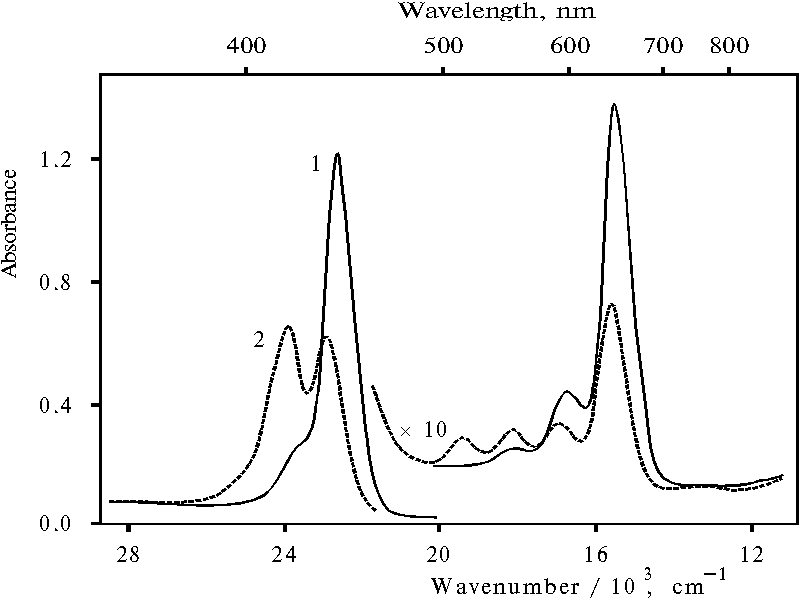
Fig. 2. Absorption spectra of meso-tetra(p-aminophenyl)porphine bound with poly-(methacrylic acid) in water, 1; and under titration with KOH at 7.85 pH, 2.
Further alkaline titration of porphyrin bound with polymer does not affect on the shift of the latter maxima. Figure 3 (curve 1) shows the titration curve of protonated porphyrin bound with polymer in the case of measurement of porphyrin concentration by the absorption at 649 nm. The plot of equation of equilibrium deprotonation in coordination of decimal log([TAPP]/[protonated TAPP]) versus pH (curve 2) gives slope of 0.99 � 0.08 that means the spectrum with the maxima at 439 and 649 nm is really singly protonated monomer of porphyrin bound with polymer.
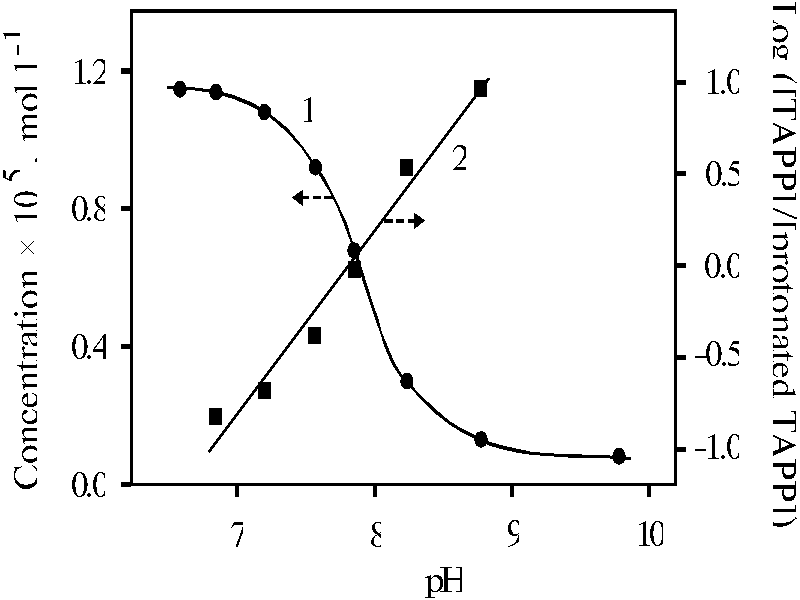
Fig. 3. The spectrophotometric titration curve of meso-tetra(p-aminophenyl)porphine bound with poly-(methacrylic acid) in water with KOH, 1; and the plot of decimal log([TAPP]/[protonated TAPP]) versus pH, 2.
At the same time, equilibrium constant of dimerization of protonated TPP in solution calculated for two cases was proved almost identical [17]. The one case took into account only concentration of different species of porphyrin while the other did concentration of water additionally with the formation of a hydroxonium ion. On the one hand, coincidence of values of the dimerization constants calculated by different way suggests competence of involvement of water molecules with formation of the hydroxonium ion. On the other hand, the coincidence means that the formation of the hydroxonium ion is not a limitation stage. Therefore, the data appears to be evidence the loss of protons by doubly protonated porphyrin molecules in the process of the charged porphyrin dimerization. Moreover, the close values of maximum of Soret bands in the spectra (Fig. 1, curve 2, lmax = 437 nm) and (Fig. 2, curve 2, lmax = 439 nm) suggest similar charge state of porphyrin molecules.
Hence, the above data complement the argumentation for the assignment of the spectrum presented in Fig. 1, curve 2 to doubly protonated dimer of tetraphenylporphine.
Fluorescence spectra of monomeric TPP and doubly protonated dimer of TPP strongly differ each from other, Fig. 4. The spectrum of TPP exhibits two bands with the maxima at 655 and 718 nm (curve 1), whereas the spectrum of the protonated TPP dimer exhibits a wide emission band with the maximum at 693 nm and a long tail in the near IR region (curve 2). It is interesting that the spectrum of porphyrin bound with polymer, where more than 90% of the bound porphyrin is present in monomeric state, exhibits a broad emission band in the near IR region with the maximum at about 1000 nm in addition to the bands with the maxima at 655 and 718 nm (curve 3). The near IR emission band appears to be due to small amount of porphyrin dimers which are present in this sample [18]. If that is the case, therefore, the porphyrin dimers are proved to be an effective traps of the excitation energy from monomeric porphyrin. As evidenced earlier, the near IR emission with the maximum at about 1000 nm originates from dimeric species of porphyrin protonating in the excited state [16, 18]. This finding looks like attractively for the study relationships and interactions between different dimeric forms of porphyrin from the point of view of excitation trapping by porphyrin dimers. In connection of this, the ground and excited states of different dimeric forms of tetraphenylporphine will be considered below.
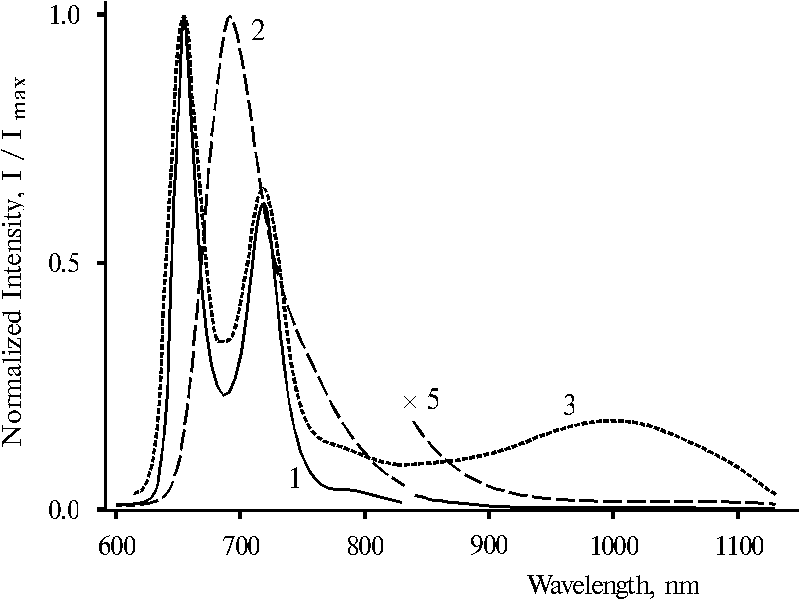
Fig. 4. Fluorescence spectra of meso-tetraphenylporphine in dioxane, 1; and in 50% aqueous solution of dioxane (v/v) in the presence of 0.2 N hydrochloric acid, 2; and meso-tetra(p-aminophenyl)porphine bound with poly-(methacrylic acid) [18] in dimethylformamide containing 1-3% of water, 3 (in this case spectral slits were some wider than in all other cases). The spectra are normalized on the intensity of the main emission band.
Figure 5 shows absorption spectra of protonated species of TPP forming in the presence of a large amount of water in organic solvent. In the UV and blue region, the spectra have three bands with the maxima at 403, 437 and 465 nm. The bands with the maxima at 437 and 654 nm appears to be characterize the doubly protonated TPP dimer. The other bands with the maxima at 465 and 694 nm are similar to those of singly protonated porphyrin dimer [18]. The spectrum of the latter is somewhat close to that of singly oxidized meso-tetra(p-aminophenyl)porphine in solution, although the red band in the spectrum of the aminoporphyrin is some red shifted because of apparently additional association of porphyrin after oxidation. In the different to doubly protonated dimer of TPP, the stability of the form with the maximum at 403 nm and a shoulder at 638 nm (see Fig. 5) was proved similar to that of the form with the maximum of the bands at 465 and 694 nm [17], i.e. the stability of the forms with the same charge is proved to be similar. Therefore, the form with the maximum at 403 nm and a shoulder at 638 nm was assigned to another singly protonated porphyrin dimer. In the difference from the region of Soret bands, the bands in the red region of the spectra are strongly overlapped that provides a base for interaction between these different dimeric forms of porphyrin.
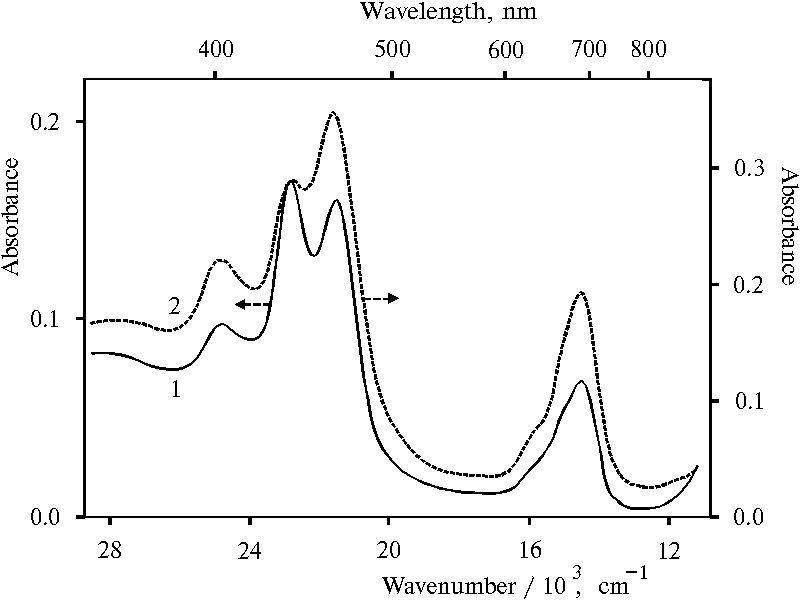
Fig. 5. Absorption spectra of meso-tetraphenylporphine in water-acetone-dioxane solution (92.5 : 5 : 2.5) in the presence of 0.4 N hydrochloric acid, 1; and in water-tetrahydrofuran solution (95.5 : 4.5) in the presence of 0.4 N hydrochloric acid, 2.
Fluorescence spectra of protonated dimeric forms of TPP presented in Fig. 6 are appreciably changed with the change of the wavelength of excitation. On selective excitation (lex = 403 and 437 nm) maximum of the main emission is observed at 693 nm (curve 1 and 2). This emission band is similar to that of doubly protonated dimer of TPP (see Fig. 4, curve 2). But the fluorescence spectrum with lex = 465 nm exhibits a maximum at 716 nm and a shoulder at 693 nm (curve 3). This effect can be seen more clear in the case of analogous water-terahydrofuran solution when different emission bands are observed more pronounced, Fig. 7. Under excitation with 403 and 437 nm, two maxima or maximum and a shoulder of the main emission are noted at 693 and 730 nm in the spectra (curves 1 and 2). While the spectrum with lex = 465 nm exhibits only one maximum at 732 nm and two shoulders at 693 nm and about 800 nm (curve 3). In the difference from the emission bands presented in the spectra (Fig. 6), these wide emission bands with the maxima at 693 and 730 nm (Fig. 7) appears to possess electron transitions from two different fluorescent states to the ground state. The energy gap between these two states estimated according to the maxima is 730 cm-1.
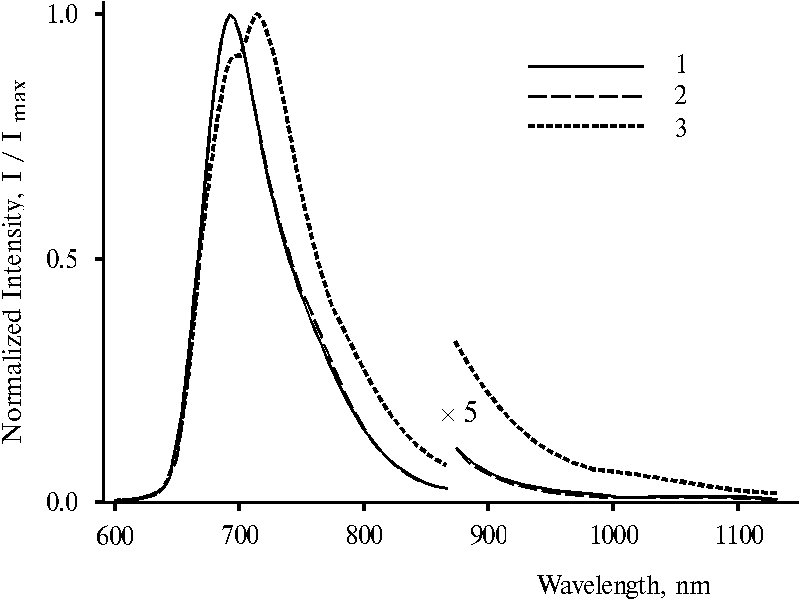
Fig. 6. Fluorescence spectra of meso-tetraphenylporphine in water-acetone-dioxane solution (92.5 : 5 : 2.5) in the presence of 0.4 N hydrochloric acid with excitation at 403 nm, 1; 437 nm, 2; and 465 nm, 3. The spectra are normalized on the intensity of the main emission band.
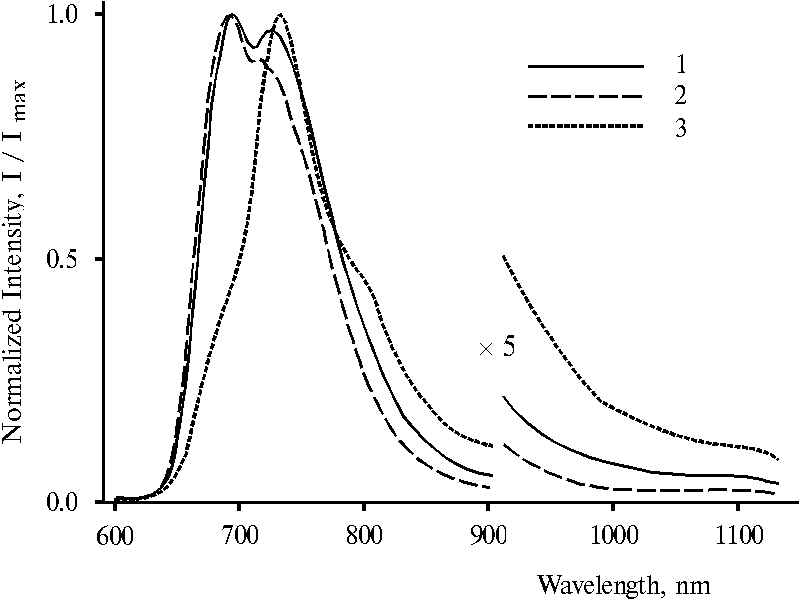
Fig. 7. Fluorescence spectra of meso-tetraphenylporphine in water-tetrahydrofuran solution (95.5 : 4.5) in the presence of 0.4 N hydrochloric acid with excitation at 403 nm, 1; 437 nm, 2; and 465 nm, 3. The spectra are normalized on the intensity of the main emission band.
Hence according to the fluorescence spectra, different protonated dimeric forms of TPP reveal different fluorescent states when there is a mixture of the forms in the solution. However, only one fluorescent state is observed in the spectrum in the case of the presence of doubly protonated TPP dimer in the solution (see Fig. 4, curve 2). Therefore, in the case of the mixture of dimers, interaction between different dimeric forms of porphyrin takes place in the excited state that is accompanied by the transfer of the excitation energy.
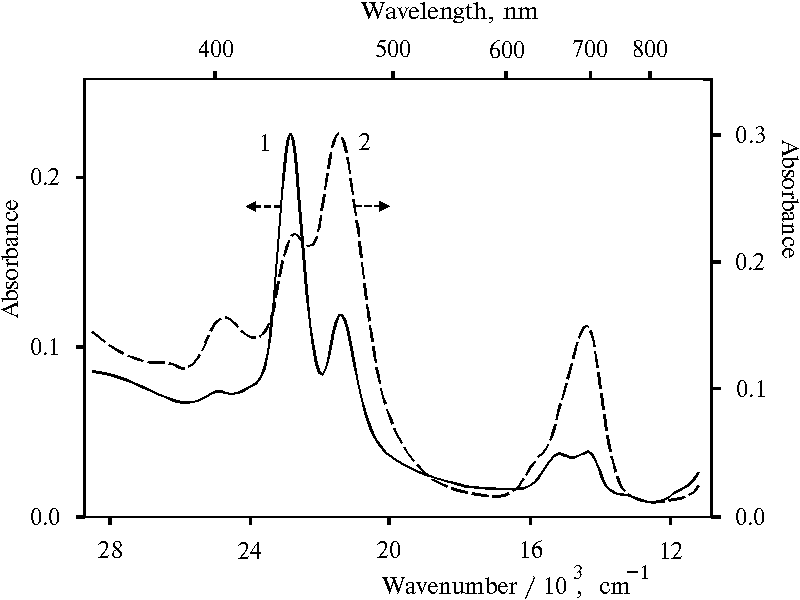
Fig. 8. Absorption spectra of meso-tetraphenylporphine in water-ethanol solution (91 : 9) in the presence of 0.4 N hydrochloric acid, 1; and in water-glycerol-tetrahydrofuran solution (86.5 : 10 : 3.5) in the presence of 0.4 N hydrochloric acid, 2.
Figure 8 shows absorption spectra of similar protonated dimeric forms of TPP prepared in the other water-organic solutions. The spectrum of the TPP dimeric forms in water-ethanol solution exhibits small and medium bands with the maxima at 403 and 654 nm, respectively, as compared to the band with the maximum at 437 nm (curve 1). The decreasing of the absorption of the 465 nm band and corresponding 694 nm band leads to the appearance of the pronounced 654 nm band which corresponds to doubly protonated TPP dimer as well as 437 nm band. In contrast, the spectrum of protonated dimeric forms of TPP in water-tetrahydrofuran-glycerol solution (curve 2) is very similar to that in water-tetrahydrofuran solution (see Fig. 5, curve 2), although maxima of Soret and red bands are red shifted by 2-4 nm in the spectrum (Fig. 8, curve 2).
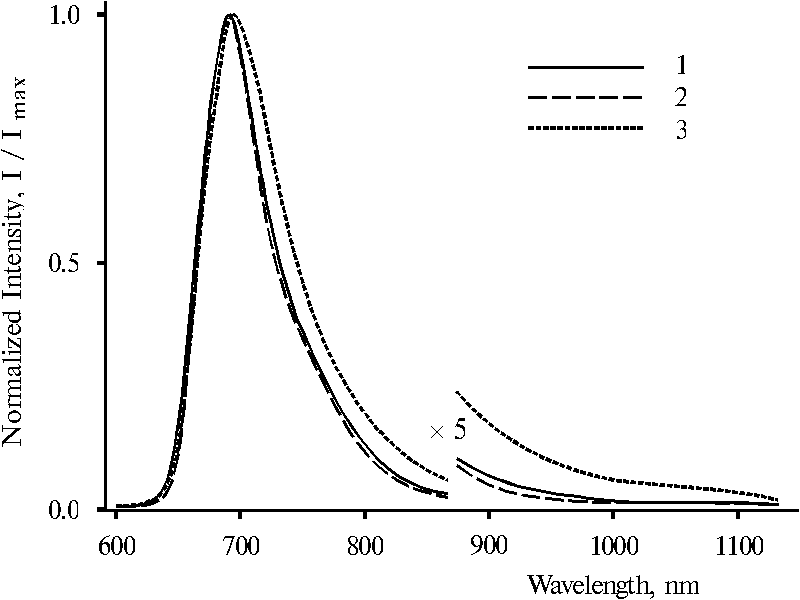
Fig. 9. Fluorescence spectra of meso-tetraphenylporphine in water-ethanol solution (91 : 9) in the presence of 0.4 N hydrochloric acid with excitation at 403 nm, 1; 437 nm, 2; and 465 nm, 3. The spectra are normalized on the intensity of the main emission band.
It is interesting that fluorescence spectra of these dimeric forms in water-ethanol solution under the same selective excitation (lex = 403, 437 and 465 nm) are very similar, Fig. 9. The spectra with lex = 403 and 437 nm are practically identical (curves 1 and 2) although, the spectrum with lex = 465 nm exhibits some broadened band, the maximum of which is red shifted by 4-5 nm (curve 3). In contrast, fluorescence spectra of protonated dimeric forms of TPP in water-tetrahydrofuran-glycerol solution presented in Fig. 10 are changed with the change of wavelength of excitation. The features of the spectra are alike to those in the case of protonated dimeric forms of TPP in water-tetrahydrofuran solution. Selective excitation (lex = 403 and 437 nm) reveals maximum of the main emission at 693 nm and a shoulder in the 700-760 nm region in the spectra (curves 1 and 2). However, the spectrum with lex = 465 nm exhibits the maximum at 730 nm and a shoulder at 693 nm (curve 3). Note that in the both cases (Fig. 7 and Fig. 10), the 403 nm excitation produces more pronounced shoulder or maximum at 730 nm in the fluorescence spectra as compared to the excitation with 437 nm.
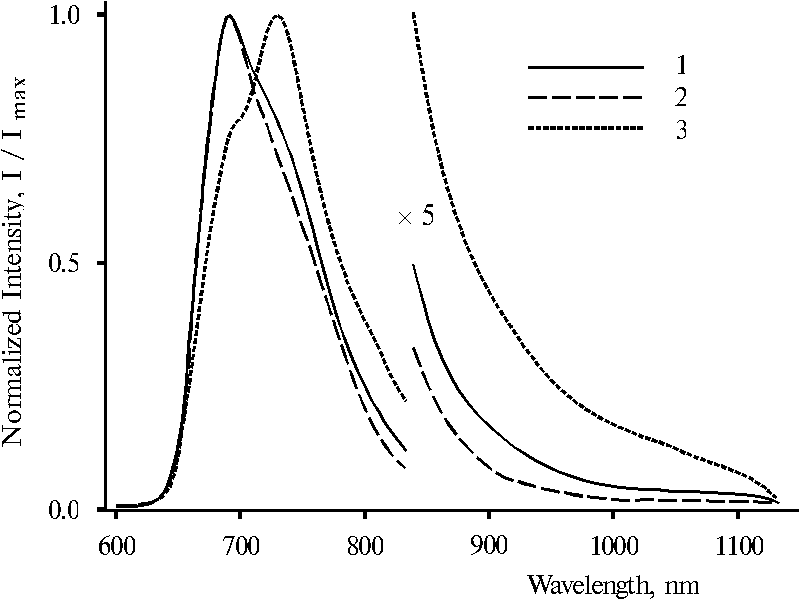
Fig. 10. Fluorescence spectra of meso-tetraphenylporphine in water-glycerol-tetrahydrofuran solution (86.5 : 10 : 3.5) in the presence of 0.4 N hydrochloric acid with excitation at 403 nm, 1; 437 nm, 2; and 465 nm, 3. The spectra are normalized on the intensity of the main emission band.
Hence, the results presented above show that the three different protonated dimeric forms of TPP observed in the ground state reveal only two different dimeric forms under transition in the excited state. The appearance of the shoulder with the maximum at about 730 nm in the spectra in the case of lex = 403 or 437 nm suggests that the fluorescence with the maximum at 693 nm can be effectively absorbed by the low-energy dimeric form of porphyrin, the absorption maximum of which is located at 694-698 nm.
Discussion
The most interesting results according to the previous section concern singly protonated dimer of TPP. It is known that under usual conditions p-electron structure of porphyrin monocation is unstable [20] but in the case of singly protonated dimeric form of porphyrin, the stabilization of the p-electron structure in the ground state occurs due to coordination of neighboring porphyrin molecule via two water molecules with the formation of hydrogen bonds [21]. As a result of the formation of hydrogen bridges between neighboring porphyrin molecules, these two water molecules can be involved into donor-acceptor interaction with porphyrin and as a consequence water-porphyrin dimeric complex can be produced. Hence, p-electron structure of the configuration of the singly protonated porphyrin dimers is stabilized in the ground state. However, in the excited state this p-electron structure is destabilized. Generally speaking, there are only two possibilities for the p-electron structure of the dimeric monocation of porphyrin to be stabilized in the excited state. In dependence from the configuration of the porphyrin dimer, a water molecule bound with the dimer can lend a proton or an electron to one of the porphyrin molecule. Therefore, we propose that p-electron structure of porphyrin macrocycle in the excited state is stabilized by lending of proton from the bound water molecule in the one configuration or electron in the other configuration of singly protonated porphyrin dimer.
The results presented in this work can be considered in the terms of this theoretical suggestion. According to the fluorescence spectra (Fig. 10), the properties of the dimeric form with the maximum at 403 nm in the excited state are similar to those of the form with the maximum at 437 nm, i.e. doubly protonated porphyrin dimer. Therefore, the bound water molecule lend a proton to this dimeric form (with the maximum at 403 nm) in the excited state. Maximum of the main emission is observed in the spectrum for this form at 693 nm (curve 1) and for doubly protonated porphyrin dimer too both in the mixture (curve 2) and alone in solution (Fig. 4, curve 2). Hence, the other singly protonated porphyrin dimer (with the maximum at 465 nm) appears to be the form where bound water molecule lend an electron to porphyrin molecule in the excited state. Maximum of main emission is observed in the spectrum for the form at 730 nm (Fig. 10, curve 3), therefore this excited state is lower by 730 cm-1 as compared to that of the former. The form with the 730 nm emission band is the interesting state because of the porphyrin dimer appears to be in the neutral state but at the same time the whole water-porphyrin complex is photochemically activated. On the one hand, this behavior of the singly protonated dimeric form can be the base for involving of water into next multi-steps oxidation process. And on the other hand, possible reaction of electron transfer between these two different singly protonated dimeric forms can be considered as a driven force in directed electron transfer in photosynthesis when the dimeric forms of porphyrin are mimicking the relationship between porphyrin pigments of photosystems I and II.
Fluorescence spectra (Fig. 7) in contrast with the spectra (Fig. 9 and Fig. 4, curve 2) suggest that strong interactions between different dimeric forms of TPP takes place when the conditions are favorable. The presence of ethanol in the solution almost prevents the emission band with the maximum at 730 nm in the spectrum (Fig. 7, curve 3), nevertheless there are different dimeric forms of TPP in the mixture. It is very probably that influence of ethanol on charged porphyrin molecules in dimer is somewhat similar to the action of methanol on cation radical of porphyrins. Addition of methanol to dication of porphyrin caused formation of isoporphyrin, i.e. tautomeric form of porphyrin, when methanol reacted with a meso carbon atom of porphyrin macrocycle [8]. Furthermore, methanol prevented from dimerization of cation radicals in contrast of other solvents (dichloromethane, chloroform) as the solution of porphyrin cation radical was cooled [5]. In this case partially resolved five lines was observed in the EPR spectrum and assigned to four hydrogens at the meso positions. Therefore, methanol and apparently ethanol as the weak agent can probably prevent from interaction of porphyrin with water. From this point of view the results presented in Fig. 9 can be understood since water molecules in this case can not lend a proton or an electron to dimeric porphyrin molecule. This prevention brings about almost similar fluorescence spectra under different selective excitation (curves 1-3).
In contrast, fluorescence spectra (Fig. 7 and 10) show different emission bands with the maxima at 693 and 730 nm where the intensity of the emission at 730 nm is strongly changed upon the conditions. A shoulder at 730 nm is only noted in the spectrum (Fig. 10, curve 1) in the presence of glycerol in the solution, while the maximum at 730 nm is observed in the spectrum (Fig. 7, curve 1) in the absence of glycerol. Hence, the increase of the viscosity of the environment considerably decreases the ability of the low-energy dimeric form of TPP (the form with lmax = 465 and 694 nm) in the trapping of excitation energy from the high-energy dimeric form (for instance, the form with lmax = 403 nm and shoulder at 438 nm). It should be noted that according to the maxima of the emission at 693 and 730 nm (Fig. 7), any selective excitation (lex = 403 or 437 nm) results in the fluorescence of both different fluorescent states likely due to the interaction between these different forms and the low-energy dimeric form of TPP. Although, the interaction between different singly protonated dimeric forms (lmax = 403 nm and lmax = 465 nm for Soret band) is proved to be some more effective as compared with the interaction between doubly protonated (lmax = 437 nm for Soret band) and the low-energy singly protonated dimeric forms, compare the intensity of the emission at 730 nm (curves 1 and 2). This fact can suggest the presence of some synchronization in the generating of the different charges in the high- and low-energy singly protonated dimeric forms of porphyrin in the excited state.
Conclusion
The data presented in this work show three protonated dimeric forms of TPP, one of which is doubly protonated porphyrin dimer (lmax = 437 and 654 nm). Two other forms are singly protonated dimers (lmax = 465 and 694 nm) and (lmax = 403 and 638 nm), the configuration of which apparently differ by orientation of dipole moments. In the red region of the absorption spectrum these three bands are strongly overlapped and the bands at 654 and 638 nm often look like in the spectra as a shoulders. Selective excitation of the dimeric forms of porphyrin (lex = 403, 437 or 465 nm) appears to reveal only two different fluorescent states of the TPP dimers. The corresponding maxima of the emission are located at 693 and 730 nm in the spectra. The excited state with the maximum of main emission at 693 nm is identified as doubly protonated porphyrin dimer. While the other state with the maximum of the emission at 730 nm is rather unstable but can be stabilized in favorable conditions. This state has probably relation to electron transfer inside water-porphyrin dimeric complex. The results presented in this work can promote the understanding of physico-chemical mechanisms of photosynthesis.
Acknowledgements
This work is supported by the Russian Foundation for Basic Research, grant No. 97-04-48155.
References
- Photosynthetic Oxygen Evolution, H. Metzner (ed.), Academic Press, London 1978.
- J.R. Darwent, P. Douglas, A. Harriman, G. Porter and M.-C. Richoux, Metal phthalocyanines and porphyrins as photosensitizers for reduction of water to hydrogen, Coord. Chem. Rev., 44 (1982) 83-126.
- Photosynthesis. Chemical models and mechanisms, ed. by V.M. Cherkasov, Nauk. Dumka, Kiev 1989, pp. 227 (in Russian).
- V.L. Pecoraro, M.J. Baldwin, and A. Gelasco, Interaction of manganese with dioxygen and its reduced derivatives, Chem. Rev., 94, (1994) 807-826.
- J. Fajer, D.C. Borg, A. Forman, D. Dolphin, and R.H. Felton, p-Cation radicals and dications of metalloporphyrins, J. Am. Chem. Soc. 92 (1970) 3451-3459.
- J.J. Katz, L.L. Shipman, T.M. Cotton, and T.R. Janson, in: The Porphyrins, ed. D. Dolphin, Chlorophyll Aggregation: Coordination Interactions in Chlorophyll Monomers, Dimers and Oligomers, Academic Press, New York 1979, vol. 5, p. 401.
- S. Gentemann, C.J. Medforth, T.P. Forsyth, D.J. Nurco, K.M. Smith, J. Fajer, D. Holten, Photophysical properties of conformationally distorted metal-free porphyrins. Investigation into the deactivation mechanisms of the lowest excited singlet state, J. Am. Chem. Soc. 116 (1994) 7363-7368.
- D. Dolphin, R.H. Felton, D.C. Borg, J. Fajer, Isoporphyrins, J. Am. Chem. Soc. 92 (1970) 743-745.
- E.I. Zenkevich, A.M. Shulga, A.V. Chernook, G.P. Gurinovich, Spectral peculiarities of NH-tautomerism in isocycle-containing porphyrins and their covalently linked dimers, Chem. Phys. Lett. 109 (1984) 306-311.
- S. Gentemann, S.H. Leung, K.M. Smith, J. Fajer, D. Holten, Photophysical consequences of porphyrin tautomerization. Steady-state and time-resolved spectral investigations of a Zinc isoporphyrin, J. Am. Chem. Soc. 99 (1995) 4330-4334.
- A.V. Udal'tsov, Absorption and luminescence spectroscopy of restrictively protonated dimeric forms of porphyrins, Biochemistry (Moscow), 62 (1997) 1026-1033.
- V.Z. Paschenko, V.V. Gorokhov, N.P. Grishanova, E.A. Goryacheva, B.N. Korvatovsky, P.P. Knox, N.I. Zakharova, A.B. Rubin, The influence of structural-dynamic organization of RC from purple bacterium Rhodobacter sphaeroides on picosecond stages of photoinduced reactions, Biochim. Biophys. Acta 1364 (1998) 361-372.
- A.D. Adler, F.R. Longo, J.D. Finarelli, J. Goldmacher, J. Assour, and L. Korsakoff, A simplified synthesis for meso-tetraphenylporphin, J. Org. Chem., 32 (1967) 476.
- A. Gordon, and R. Ford, Sputnik Khimika, Mir, Moscow, 1976, p.437 (in Russian).
- A.A. Krasnovsky (Jr.), Photoluminescence of singlet oxygen in pigment solutions, Photochem. Photobiol., 29 (1979) 29-36.
- A.V. Udal'tsov, V.Z. Paschenko, A.A. Churin, V.B. Tusov, V.S. Pshezhetskii, Donor-acceptor interactions in porphyrin associates immobilized in biphilic copolymer J. Photochem. and Photobiol. B: Biol., 21, (1993) 87-94.
- A.V. Udal'tsov, Absorption and luminescence spectroscopy of restrictively protonated dimeric forms of porphyrins, Biochemistry (Moscow), 62 (1997) 1026-1033.
- A.V.Udal'tsov, Characteristics of donor-acceptor complexes formed in porphyrin-polymer systems and their photoactivation in electron transfer photoreaction. J. Photochem. and Photobiol. B: Biol., 37 (1997) 31-39.
- V.S. Pshezhetskii, A.V. Udal'tsov, Influence of modified poly(methacrylic acid) covalently bound to porphyrin on its acid-base properties, Vysokomolek. Soedin., A30 (1988) 1470-1475 (in Russian).
- P. Hambright and E.B. Fleischer, The Acid-Base Equilibria, Kinetics of Copper Ion Incorporation, and Acid-Catalyzed Zinc Ion Displacement from the Water-Soluble Porphyrin a,b,g,d, -Tetra(4-N-methylpyridyl)porphine, Inorg. Chem., 9 (1970) 1757-1761.
- A.V. Udal'tsov, A.A. Churin, Molecular complexes between dimeric forms of porphyrin and water and their vibrational dynamics, Internet Photochem. Photobiol., (1998) http://www.photobiology.com/IUPAC98/Udaltsov/index.htm