| Papers and Posters | Site Home Page |
FLUORESCENCE LABELED DNA PROBES FOR HOMOGENEOUS DETECTION OF COMPLEMENTARY SEQUENCES
Eva M. Talavera1; Moustafa Afkir1; Rafael Salto2; Alberto M. Vargas2 and Jose M. Alvarez-Pez1*
Departments of Physical Chemistry1 and Biochemistry and Molecular Biology2.
Facultad de Farmacia. Universidad de Granada. Campus de Cartuja s/n. 18071 Granada
(Spain).
*To whom correspondence should be addressed.
Abstract
The development of new sensitive, safe and easy to use probes for the detection of nucleic acids is of relevant importance. We are trying to develop a fluorescence based technique to work in homogeneous assay systems. Two different labels have been used, pyrene and fluorescein. In this work we are presenting results of the change in steady-state fluorescence intensity occurring when a single-stranded labeled cDNA is hybridized with the complementary strand. Our results suggest that pyrene based labeling is more promising than fluorescein to detect and quantify DNA from natural sources.
Introduction
Identification as well as quantification of particular DNA sequences in complex DNA preparations is of remarkably interest in various fields, including among others, basic research, diagnosis of infectious and genetic diseases, forensic medicine and many biotechnological applications.
The presence of particular sequences may be quantified by the use of PCR in order to amplify that sequence, if present. Other general approach is the use of specific DNA or RNA probes. At present, radioactive probes are the most efficient, although many non-radioactive labeling techniques are in development in order to avoid the use of radioactive materials. An important drawback of these techniques is that most of them, including those using radioactivity, require complex separation steps to remove free from hybridized probe. These are called heterogeneous assays. In other cases, an indirect method is used for detection, thus requiring additional incubations with the secondary agent containing the detecting label. In contrast, homogeneous assays are simpler, not requiring further treatment for the detection of the amount of hybridized probe. Fluorescence techniques using fluorescent labeled DNA probes have the potential for developing homogeneous, relatively inexpensive, and easy to use DNA probe assays, due to the sensitivity of the emission intensity of the fluorescent label to environmental changes. Such assays are possible if the hybridization of the probe with the target sequence is accompanied by a change in one or more fluorescent properties such as fluorescence quantum yield, lifetime, polarized emission, fluorescence quenching, excitation transfer or sensitized fluorescence. The target sequence can thus be detected and quantified from the change in properties occurring when the target sequence is added to an analysis tube containing the appropriate DNA probe. So far, there are only a few works reporting fluorescent based hybridization detection schemes in homogeneous media. Cantor et al. [1] have reported studies in which the 3' terminus of single-stranded RNAs were labeled with pyrene. Titration of pyrene-labeled poly(C) with unlabeled poly(I) resulted in a decrease in the fluorescence intensity by a factor of about 2 at saturating concentrations of poly(I). However, the graph of fluorescence intensity versus concentration of poly(I) was complex and did not vary linearly with poly(I) concentration. Methods for implementing a homogeneous DNA assay based on excitation energy transfer have been described. Morrison and Stols [2] described a method using two DNA probes, one of them labeled with a donor group attached to the 3' and the other one labeled with an acceptor group attached to the 5' end. The two probes must be designed in such a way that when they bind to the target sequence, the donor and acceptor groups are next to each other. Recently, Yguerabide and Ceballos [3] have described the use of intercalating fluorophores for determining DNA in homogeneous assays. We have described in a previous paper a synthetic model system, in which we used a pyrene labeled polyribocytidilic acid (5'), poli (C), as DNA probe and polyriboinosinic acid (5'), poly (I), as a target in an homogeneous assay [4]. We have also developed fluorescent techniques in homogeneous media to measure DNA melting and the rate of renaturation by the use of fluorescein attached to fragments of E. coli DNA for the detection of the fluorescence change when double stranded-DNA was denatured [5].
The experiments described in this paper were designed to verify the utility of the
previously described methods in a homogeneous system formed by a fluorescent-labeled
single stranded-cDNA (ss-cDNA) probe from an eukaryotic gene widely used in the study of
gene expression when it is allowed to anneal with the ss-cDNA unlabeled complementary
strand. Implementation of the method presented here will allow, with a very easy
homogeneous method, the detection and quantification in any DNA sample of the
complementary sequences to previously fluorescent labeled probes.
Materials and methods
Reagents. Fluorescein isothiocyanate, sodium bisulfite, ethylene diamine, Tris [hydroxymethyl] aminomethane (Tris), 2-hydroxy-ethylpiperazine-2-ethanesulfonic acid (HEPES), sodium chloride, dimethylsulfoxide (DMSO), monosodium and disodium phosphates were obtained from Sigma Chemical Company. Pyrenesulfonyl chloride was from Molecular Probes, Inc. Other reagents for molecular biology were from Pharmacia or Promega. All reagents were of maximum purity and used without further purification.
Cloning of cDNA. We have used for all the experiments in this paper a fragment of 428 bp from the glyceraldehide 3-phosphate dehydrogenase (GA3PDH) gene, obtained from a rat-liver cDNA library [6]. A set of two oligonucleotides with the following sequences GAPDH-Forward (5'- CCCACGGCAAGTTCAACGG-3' Tm= 62ºC) and GAPDH-Reverse (5'-CTTTCCAGAGGGGCCATCCA-3' Tm=64ºC) have been used to amplify the cDNA by a standard PCR [7]. The reaction was carried out in 50 µl of a solution containing 50 pmol of each primer, 200 µM each of dATP, dGTP, dTTP and dCTP, template DNA, 10 mM Tris-HCL, pH 8.3, 50 mM KCl and 1.5 mM MgCl2 in 0.5 ml eppendorf tubes. The 428 bp obtained fragment has been isolated by electroelution after electrophoresis purification and cloned in a pGEM-T vector from Promega. The orientation of the fragment has been determined after Nco-I digestion, taking advance of an internal restriction site and the one in the polylinker of the vector.
Preparation of single stranded cDNA (ss-cDNA). The plasmid was linealized either with Apa-I or with Pst-I in order to set a run-off PCR polymerization technique [8]. To get the forward strand the construct was linealized with Pst-I and GA3PDH-F was used as primer. The reverse strand was obtained after digestion with Apa-I using GA3PDH-R as primer. In both cases, the asymmetric PCR was conducted with 200 pmol of primers in 50 µl final volume during 45 cycles. After electrophoresing the samples in an agarose gel the single stranded amplified bands were clearly visible with ethydium bromide allowing the electroelution of the two separate ss-cDNAs.
ss-cDNA labeling. Using the Shapiro and Weigras technique [9], we have replaced the cytosine amino group at the N4 position by other amine-containing small organic molecule. This modification introduces a derivatized free amino group with a spacer arm where further addition of fluorescent labels can be attained by using fluorophores with amine specific reagent groups. The first stage was conducted in a water solution containing 3 M ethylenediamine and 1 M sodium bisulfite prepared immediately before labeling experiments. One volume of the forward strand ss-cDNA (1 mg/10 ml) suspended in 10 mM Tris-HCl, 1 mM EDTA, pH 7.5 buffer, was added to nine volumes of the ethylenediamine-sodium bisulfite solution and heated at 42ºC during 1 hour, to achieve a modification of around 4% of the total nucleotide bases. 50 ml of 200 mg/ml hydroquinone dissolved in 95% ethanol was added to scavenge free radicals. The modification reaction was stopped by placing the reaction tube on ice and adding 100 ml of 1.0 M HEPES buffer pH 10.0. To obtain the N4-ethylenediamine substituted cytosine residues and remove the bisulfite adduct, DNA solution was dialyzed against 20 mM phosphate buffer pH>8.5 overnight, with at least three changes of buffer. Modified ss-cDNA was concentrated from the dialysis bags by precipitation with ethanol and NaCl addition.
The DNA thus modified has been employed for labeling with either fluorescein isothiocyanate according to Reines and Schulman [10] or pyrenesulphonyl chloride according to Liu et al. [11].
In a typical fluorescein labeling experiment, 1 mg of DNA was suspended in 20 ml of 5 mM sodium phosphate buffer pH 7.5 in an eppendorf tube, supplemented with 10 ml of 1.0 M Hepes buffer pH 10.0 to maintain pH > 8.0 as the hydrolysis of free fluorophore isothiocyanates can significantly lower the pH. A 10 mg/ml solution of fluorescein isothiocyanate in DMSO was prepared immediately before use. An equal volume of the fluorophore solution was then added and the reaction mixture incubated for one hour at 37ºC. Labeled nucleic acids were purified by repeated precipitation with ethanol until there was no detectable fluorescence from free fluorescein in the remaining supernatant. The last step for purification of the labeled material was by gel permeation on Sephadex G-25. When ss-cDNA was labeled with pyrenesulfonyl chloride, 1 mg of modified ss-cDNA was suspended in 200 ml of 5 mM sodium phosphate buffer pH 7.5 in an eppendorf tube. 100 ml of 1.0 M Hepes buffer pH 10.0 were added subsequently. Next, 100 ml of a 6 mg/ml pyrenesulfonyl chloride in acetone solution prepared immediately before labeling, were added dropwise for 1 h at 37 ºC. The solution was purified from free pyrene by repeated steps in Sephadex G-25 gel permeation column. After each gel permeation step the solution was precipitated with ethanol and tested for pyrene emission in the remaining supernatant. This purification step was repeated until there was no detectable fluorescence.
Determination of the extent of labeling. The extent of labeling was determined on strands of labeled ss-cDNA by absorption spectroscopy. To determine the actual dye-to-nucleotide base ratio, one must subtract the absorbance of the fluorophore at 260 nm from the total 260 nm absorbance. The following equation applies:
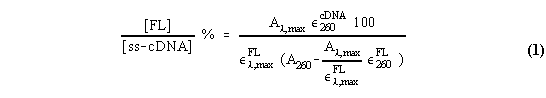
where lmax is the wavelength at the maximum absorption of the fluorophore, FL; Al,max and A260 are the absorbances at lmax and 260 nm, respectively; el,maxFL, e260cDNA and e260FL are the decadic extinction coefficients of fluorophore at lmax, of cDNA at 260 nm and of the fluorophore at 260 nm, respectively. Equation (1) gives the labeling ratio as the molar concentration of fluorophore to the concentration of ss-cDNA expressed as molar concentration of nucleotide bases. The molar absorption coefficients used were those previously described [4,5]. For ss-cDNA a e260DNA of 10,000 M-1 cm-1 was used. Determination of the amount of fluorophore by hydrolyzing samples and releasing the fluorophore by incubation of the labeled material in 0.3 M NaOH, produces similar results [4, 5].
Spectral measurements. The absorption spectra were recorded using a Perkin-Elmer Lambda 5 spectrophotometer equipped with a water jacketed cuvette holder. Fluorescence spectra were recorded with a Shimadzu RF-5001 spectrofluorometer equipped with a controlled temperature cell holder with stirring system. For pyrene detection, excitation was performed at 350 nm and changes in fluorescence emission were recorded between 370 and 500 nm. For fluorescein detection, excitation was at 480 nm and emission between 495 and 550 nm.
In hybridization experiments, the labeled strand dissolved in 20 mM Tris-HCl, 0.1 M
NaCl, 1 mM EDTA, pH 7.5 (hybridization buffer) in the presence of either 50 % formamide or
50 % ethanol was temperature controlled in the cuvette of the fluorometer. To this
solution the complementary strand was added and mixed quickly and spectra were taken along
time.
Results and Discussion
Pyrene is a hydrophobic molecule whose fluorescence efficiency increases with a decrease in solvent polarity. As we have previously shown [4] the hydrophobic pyrene molecule attached to poly(C) is expected to locate preferentially within poly(C) hydrophobic plaits. When poly(I) hybridizes with pyrene-poly(C), the pyrene label is expelled outward from the double strand because of steric hindrance and therefore the new hydrophilic environment causes both the decrease in fluorescence efficiency and the shift of the emission spectrum to red. A similar behavior can be expected for labeled natural DNA molecules.
For optimum hybridization properties, nucleic acids should be labeled, at most, to 5% of the total bases [5]. However, this percentage must be lower when pyrene is used because of the pyrene excimer formation trend, producing a characteristic emission spectrum with a maximum around 500 nm concomitantly to a decrease of the peak intensity at 385 nm, leading to a diminution of the detection sensitivity. In accordance, we have used probes with a label percentage between 2-3% of either pyrene or fluorescein.
The specificity of DNA hybridization is mostly influenced by the temperature of the assay. Thus, it is of major importance to select the appropriate conditions to ensure the maximal temperature giving an specific hybridization without disrupting the bonds of the pyrene probe.
Since the stability of the label bond is temperature dependent, hybridization must be performed below 50 ºC. Either formamide or high salt concentrations should be added in accordance. An experiment was designed to test any possible influence of formamide on the emission spectrum characteristics. A pyrene-poly(C) was hybridized with poly(I) as in [4], except that 50 % formamide was present. To ensure the annealing, temperature was maintained at 20 ºC during the experiment. Figure 2 shows the changes in fluorescence intensity which occurs when 400 ml of a 5.2 x 10-5 M solution of pyrene-poly(C), in hybridization buffer:formamide (50:50) solvent, is titrated with aliquots of 50 ml of a 7 x10-5 M solution of poly(I) in the same solvent. Fluorescence intensity decreases continuously and the spectrum shifts towards red with increasing concentration of poly(I) until saturation is reached at a molar ratio of 1:1 in nucleotide bases of poly(C) to poly(I). After each addition of poly(I), the sample was incubated for 15 min at 20 ºC before recording the fluorescence spectrum.
The steady-state fluorescence intensities at 385 nm of Figure 2, after corrected by dilution effect, decrease about 70% when poly(I) completely hybridizes with pyrene-poly(C). This change in fluorescence intensity is similar to previously published data for pyrene-poly(C)-poly(I) in hybridization buffer [4] and indicate that formamide did not perturb the titration sensitivity.
To determine the temperature of the hybridization reaction of the natural probe from GA3PDH cDNA in order to minimize nonspecific binding, we have calculated the melting temperature of the ds-cDNA by the expression [12]:
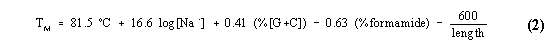
For our cDNA, length is equal to 428 base pairs and G+C is 237 = 55.4%. If [Na+] is 0.1 M, application of equation (2) gives a TM of 86 ºC without formamide and TM = 54.5 ºC in the presence of formamide. Moreover, the bimolecular rate constant of cDNA hybridization shows that the formation of duplex between complementary ss-cDNA, in hybridization buffer: formamide (50:50) solvent, after 10 hours of reaction is about 95% when initial ss-cDNA concentration is equals to 4.5 x 10-6 M [12].
We decided to use a temperature of 45 ºC for the hybridization to ensure selective and complete hybridization. Consequently, we needed to show that at this temperature the pyrene label remained attached to ss-cDNA. A pyrene-poly(C) solution was kept at 45 ºC over a time period of 10 hours. After this time, the solution was refrigerated at 20 ºC and its fluorescence recorded. A small decrease in fluorescence intensity was observed when compared to that obtained at 20 ºC at the beginning of the experiment. After the annealing of the probe with a saturating concentration of poly(I), the percentage of decrease obtained, about 66%, was of similar magnitude than that shown in Figure 2. This result means that only an insignificant amount of fluorescent label is released from the probe after 10 hours at 45 ºC.
Once the experimental conditions were set, we carried out an homogeneous hybrydization experiment using our labeled cDNA. Figure 3 shows the changes in fluorescence intensity occurring when 100 ml of a 2.25 x 10-5 M solution of ss-cDNA (reverse strand of GA3PDH) were added to 400 ml of a 5.6 x 10-6 M solution of pyrene-ss-cDNA (forward strand of GA3PDH), in hybridization buffer:formamide (50:50) solvent at 45 ºC. The cuvette was covered with a Sigma Cot solution, hydrophobic film to prevent any attachment of the probe to its walls. The fluorescence spectra were recorded over a time period of 24 hours. As shown in Figure 3 the peak fluorescence intensity decreases continuously with time during the first 10 hours of reaction, remaining constant since then. The total decrease in the steady-state fluorescence intensity at 385 nm upon hybridization is about 30%, a percentage smaller than that obtained when pyrene-poly(C)-poly(I) hybridization is observed.
Several reasons may be argued for the interpretation of this smaller decrease. Firstly, it may be explained in terms that a poorer displacement of the pyrene label to the water is produced in this circumstance due to the differences in the hybridization pattern between the two DNAs. The extent of annealing with poly-(C)-poly-(I) must be higher in this situation as any chain may hybridize with any part of the complementary and a network of interbreed strands may be produced. With natural DNA, hybridization must be produced exactly between any single chain with the complementary. A second point to consider is that pyrene-poly-(C) may not form any secondary structure while a single-stranded natural DNA may contain stretches of double helix formed by base complementarity. Using a program to detect maximum pairing in RNA structures (13) almost 50 % of the sequence could form double-helix. Although this calculation has been done with the sequence of the RNA corresponding to the insert of GA3PDH and there are differences in the hybridization constants of DNA and RNA, we can not exclude the possibility that a percentage of the label stays from the beginning of the experiment in the aqueous phase. The hybridization with the reverse strand will not consequently change any spectra properties on this molecules. Finally, the length of the forward strand utilized as the label probe is 428 bases corresponding to the GA3PDH gene followed by a 21 bases stretch corresponding to the distance from the end of the insert to the Pst-I restriction site in the polylinker of the vector. These 21 bases have no possibility for hybridization with the complementary reverse strand, which is constructed in the other direction. This fact alone explains a 5 % difference among poly-(C)-poly-(I) and the natural DNA assayed in this work.
The other fluorescent label that we have used is fluorescein. As we have established elsewhere [5], changes in the steady-state fluorescence intensity which occur when DNA labeled with fluorescein is renatured are due to changes in the fluorescein monoanion-dianion ratio producing an apparent modification in the pKa of the conjugated fluorescein when it is in a double-stranded structure. These changes are attributed to the increase in local negative charge density from the phosphate groups of the nucleic acids.
We have observed that addition of 50% formamide to the hybridization experiment gives rise to a fluorescence quenching that considerably reduces the detection sensitivity. For this reason, in the experiments with fluorescein-ss-cDNA we have used a solvent hybridization buffer:ethanol (50:50) that has similar effects than formamide in the TM value of ds-cDNA at the same temperature of hybridization[5].
Another problem using fluorescein is photobleaching produced by the repeated excitation with a xenon lamp of a small amount of sample with a short percentage of fluorescein-label. Therefore, we have performed the hybridization experiments with a 3 % of fluorescein conjugated to the forward ss-cDNA using two aliquots of 420 ml of a solution 1 x 10-5 M of the probe in hybridization buffer:ethanol (50:50). One of them was added with 80 ml of a 5.5 x 10-5 M solution of the complementary ss-cDNA in the same solvent, while the other was diluted with 80 ml of the solvent and was used to subtract photobleaching. The emission spectra between 495 and 550 nm, were recorded six times for 12 hours and a final spectrum was taken after 24 hours. The recorded spectra are shown in Figure 4.
In our sample we measured a decrease of the fluorescence intensity at 515 nm of 29 %, whilst the photobleaching control showed a 14 % of decreasing, meaning that only a 15% decrease should be assigned to the fluorescence quenching of fluorescein specific of the hybridization reaction. The percentage of decrease achieved here if labeling only one of the strands of DNA is similar to that obtained in our previously described experiments [5] in which both strands of total DNA from E. coli were labeled, showing that no improvement is made when ss-DNA is used. This makes the development of a method for detection of DNA hybridization using fluorescein difficult.
In conclusion, pyrene-ss-cDNA results in a larger decrease percentage upon
hybridization than fluorescein-ss-cDNA and allows its use as a nucleic acid probe in a
homogeneous assay. At present the fluorescence homogeneous assays described here do not
have enough sensitivity to detect very little amounts of target DNA. Sensitivity could
possibly be further increased by allowing a quicker hybridization among the labeled probe
and the complementary strand. We are at present developing several techniques in this
sense.
Acknowledgements. We deeply thank Prof. Paloma Hortelano for her critical
reading of the manuscript and for helping with the English usage.
References
[1] P. Koenig, S.A. Reines and C.R. Cantor, Pyrene derivatives as fluorescent probes of conformation near the 3' termini of polyribonucleotides, Biopolymers 16 (1977) 2231-2242.
[2] L.E. Morrison and L.M. Stols, Sensitive fluorescence based thermodynamic and kinetic measurements of DNA hybridization in solution, Biochemistry 32 (1993) 3095-3104.
[3] J. Yguerabide and A. Ceballos, Quantitative fluorescence method for continuous measurement of DNA hybridization kinetics using a fluorescent intercalator, Anal. Biochem. 228 (1995) 208-220.
[4] J. Yguerabide, E.M. Talavera, J.M. Alvarez-Pez and M. Afkir, Pyrene-labeled DNA probes for homogeneous detection of complementary DNA sequences: poly(C) model system, Anal. Biochem. 241 (1996) 238-247.
[5] E.M. Talavera, J.M. Alvarez-Pez, L. Ballesteros and R. Bermejo, Fluorescein- labeled DNA probes for homogeneous hybridization assays: Application to DNA E. coli renaturation, Appl. Spectrosc. 51 (1997) 401-406.
[6] M.D. Girón, A.M. Vargas, M.D. Suárez and R. Salto, Sequencing of two alternatively spliced mRNAs corresponding to the extracellular domain of the rat receptor for advanced glycosylation end products (RAGE), Biochem. Biophys. Res. Commun. 251 (1998) 230-234.
[7] H. Neumann, A. Cavalié, D.E. Jenne and H. Wekerle, Induction of MHC class I genes in neurons, Science 269 (1995) 549-552.
[8] M. Stürzl and W.K. Roth, Run-off synthesis and application of defined single- stranded DNA hybridization probes, Anal. Biochem. 185 (1990) 164-169.
[9] R. Shapiro and J.M. Weisgras, Bisulfite-catalyzed transamination of cytosine and cytidine, Biochem. Biophys. Res. Commun. 92 (1970) 422-424.
[10] S.A. Reines and L.H. Schulman, A new method for attachment of fluorescent probes to DNA. Methods Enzymol. 59 (1979) 146-156.
[11] B.M. Liu, H.C. Cheung and J. Mestecky, Nanosecond fluorescence spectroscopy of human immunoglobulin A, Biochemistry 20 (1981) 1997-2003.
[12] T. Maniatis, E.F. Fritsch and J. Sambrook, Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York (1988).
[13] M. Zuker, Computer Prediction of RNA Structure in RNA processing, Methods Enzymol.
180 (1989) 262-288.