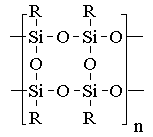
| Site home page | Conference home page | Discussion |
Photochemical crosslinking of the low molecular weight vinylcontaining polysiloxanes with organic azides
Nadezda V. Zelentsova*, Sergei V. Zelentsov*, Mark J.M. Abadie**, and Elena N.Makareeva*
* Nizhny Novgorod State University,
Gagarin Ave., 23. Nizhny Novgorod, 603600, Russia
**Universite Montpellier II, France
Keywords: linear polysiloxane, cyclolinear polysiloxane, vinyl containing oligomer, photochemical crosslinking, organic azide, nitrene, reactivity
Summary: Photochemical crosslinking of vinyl-containing polysiloxanes having linear and cyclolinear (ladder-like) structure with aromatic diazides has been studied. It was shown that the reaction involves the aziridine bridge formation, the terminating OH-groups polymers having no part in it. In cyclolinear oligotetracyclosiloxanes the crosslink formation effectiveness is greater for arylnitrene compared to the one for sulfonylnitrenes. In linear polysiloxane the former value is less than the latter one. This suggests better access “approachability” of the vinyl groups bonded with cyclolinear oligotetracyclosiloxanes towards the arylnitrenes attacks compared with the sulfonylnitrenes attacks and formation of special structures existing in polymer solutions; such structures having parallel interposition of vinyl and azidogroups towards each other. The increase of the u.v. light sensitivity of compositions containing cyclolinear oligovinyltetracyclosiloxanes against the one containing linear polysiloxanes may be explain with a “solid state“ polymerization of close packed vinyl groups initiated by nitrenes or/and radical products emerging from their reactions with double bonds.
The vinyl containing polysiloxane oligomers are the perspective microelectronic materials. One of the methods of their application is a photochemical modification with diazides. Although the organic diazides are widely spread and have a high sensitivity with respect to u.v. light their usage as the oligosiloxane photocrosslinking agents were not studied properly.
1. Introduction
The photosensitive compositions consisting of aromatic azide and polysiloxane were described in some publications. One of them contained 3-20 mol. % of azide in resp. to polymer having the average polymerization degree of 1000-10000 [1]. Other technical solution described [2] had photosensitive composition consisting of the base soluble phenolic resin and aromatic azide in weight ration 1.00:0.25-1.00 with admixture of 10-80% of bis-(1,3,5,7-tetrahydroxy)-1,3,5,7-tetraphenylcyclotetrasil-oxane. In addition the azide group can be included into the silicon containing polymer molecules [3-5]. It was described the composition which contained polyorganosiloxane having fragment
attached to silicon atom through carbon atom, this polymer contained vinyl groups, and the composition contained not more than 3% of organic peroxide. Sometimes the silylated novalac resin was used as a polymeric binder [6].
And to conclude this brief review it is necessary to mention that presence in polymer nodes containing both silicon and unsaturated bonds increased the sensitivity when used in composition with azide [7].
2. Experimental part
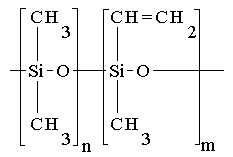
We have investigated a photocrosslinking of cyclolinear tetrasubstituted oligotetracyclosiloxane (A) and vinyl containing linear polysiloxanes (B) with diazides.
|
(A) |
(B) |
|||||||
| MW | Tg | m/n | MW | Tg | ||||
| 1: R = CH=CH2 | 10000 | 373 K | 5:0.00 | 100000 | 150 K | |||
| 2: R = CH3 | 7000 | 6: 0.04 | 100000 | 150 K | ||||
| 3: R = C6H5 | 1000 | 7: 0.07 | 100000 | 150 K | ||||
| 4: CH=CH2/CH3 = 1:3 | 8: 0.023 | 50000 | ||||||
| 9: structure (B): m=0, MW=50000; macromolecules is terminated with CH2=CH-groups. | ||||||||
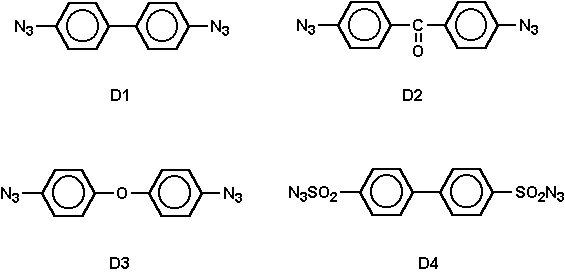
The diazides used were 4,4'-diazidobiphenyl (D1), 4,4'-diazidobenzophenone (D2), 4,4'-diazide biphenyloxide (D3), and 4,4’ -diazide disulfonylbiphenyl (D4).
The polymer and the diazide (the content of the latter being 3 w.% with respect to the polymer ) were dissolved in organic solvents (toluene and chloroform). The films were formed by coating a small amount of the solution onto the glass substrates (by means of spinning technique) and then dried for 4 h at the ambient temperature (295 K). The films were irradiated with unfiltered u.v. light of DRK-120 lamp. IR spectra were recorded with Specord 75 IR spectrophotometer.
3. Results and Discussion
The unanimous chemical mechanism of photochemical crosslinking of polysiloxanes with diazides has not been established yet. Averichkin et al [8] observed a decrease of HO vibrations and an increase of SiO-vibrations intensities in IR spectra of the oligosiloxanes being irradiated with the E-beam radiation. They proposed that the oligotetracyclosiloxanes crosslinking initiated by E-beam exposure took a way of an intermolecular dehydration of the terminal hydroxyl groups. The same mechanism might be postulated in the case of the polysiloxane photocrosslinking with diazides. To test the possibility the changes of the IR spectra of the films formed from 1-D1 system under u.v. light irradiation were recorded. We were not able to observed any noticeable increase of the option in the ranges 3700-3600 (n OH), 3600-3200 (n NH), 1200-1000 (d SiO) cm-1. However there had place the synchronous decreasing of both azide and vinyl groups vibration intensities in the ranges 2130-2074 (n as(N3)) and 1300-1200 (n s(N3)), and 1636-1616 (vinyl group vibrations) cm-1 ( Fig.1).
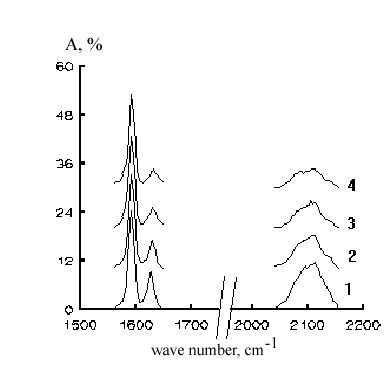
Figure 1. Changing of IR-spectra of oligosiloxane films with the u.v. irradiation intensity. Here: time of the irradiation, s, (No. of curve ): 0(1), 2(2); 10(3); 30(4).
Having this in mind, we proposed that the main event of the crosslinking process was an interaction of the photochemically generated nitrenes with the oligomer vinyl groups forming aziridines. This scenario is usually postulated for any double bond containing polymer [9]
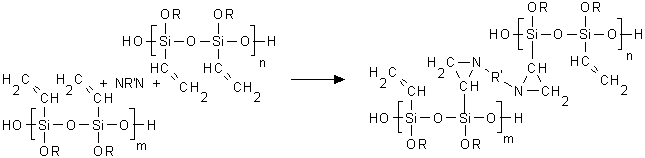 (1)
(1)
To detail the mechanism proposed we produced a study of the photochemical reaction kinetics of the diazides in the polysiloxanes. As a measure of the relative photochemical reactivity we used the minimum dose of the u.v. light exposure, DM, which are sufficient for a loss of the exposed film solubility in the organic solvent (toluene).
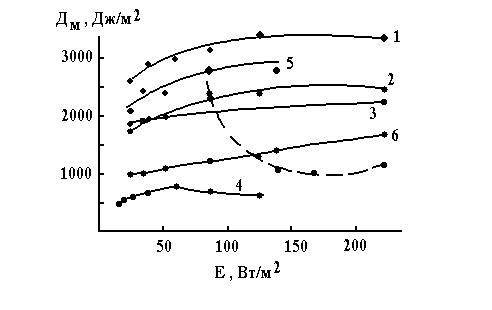
Figure 2. Dependence of DM for oligosiloxane films on the irradiation intensity in presence of oxygen. Here: 1 – 5-D1; 2 – 1-D4; 3 – 6-D1; 4 – 7-D1; 5 – 5-D4; 6 – 1-D1; 7 – cyclized polyisoprene – 2,6-di(4’-azidobenzelidene)-cyclohexanone (from [10]).
Fig.2 shows the influence of the u.v. light irradiation intensity, E, on DM of some systems exposed in presence of oxygen. Curve 7 shown as dashed line represents the influence in the case of films made of cyclized polyisoprene containing diazide D1 [10]Comparing the curves 1-6 with 7 one can easily notice that they differ in the region of small intensities. In the case of cyclized polyisoprene system DM increases greatly when E is small. In contrast to the latter system the polysiloxane systems are less sensitive to the u.v. light intensity decreasing. It is usually assumed [9,10] that the DM increasing due to the u.v. light irradiation intensity decreasing is caused by the nitrene oxidation with molecular oxygen, the latter process competing with interactions of a nitrene with polymer.
The photooxidation of diazides seems to give small contribution in the case of polysiloxanes. The latter agrees with observations made in [11]. The absence of great influence of E upon DM gives an opportunity to use DM as a measure of relative reactivity of the diazide-polysiloxane pairs. Table 1 contains DM values determined for the u.v. light intensity of 50 W · m-2.
Table 1
The solubility loss minimum dose for the polysiloxane-diazide systems (determined at 50 W· m-2 intensity of the u.v. light).
|
System |
DM, J· m-2 |
System |
DM, J· m-2 |
|
1-D1 1-D2 1-D3 1-D4 2-D1 2-D2 3-D1 3-D2 |
500
22.5 1000 3670 4800 4530 < 10000 < 9000 |
4-D1 5-D1 5-D4 6-D1 7-D1 8-D1 9-D1 |
10000 3000 1040 2670 2400 3500 2000 |
We have studied liquid (using polydimetylsiloxane) and solid (using oligotetrasubstituted cyclosiloxane 1-4) systems. In the former systems DM is controlled by the liquid phase reactivity of a nitrene depending upon an electronic charge located on the nitrogen atom [12]. This is why the sulfonylnitrene reactivity 1.7 times greater than the arylnitrene one (cf. systems 5-D1 and 5-D4). The reactivities of nitrenes in the solid state are influenced less by the electronic charges than by the structural factors such as the reagent mobilities, structural retarding of the reaction attack by a nitrene, the initial mutual positions of the reagents [13]. One can see from Table 1 that the 1-D1 system has DM being 7.5 times less then DM for the 1-D4 system. Such changes can be caused by higher "structural difficulty" for a sulfonylnitrene compared to an arylnitrene to come close to a double bond of the oligomer and produce a configuration suitable to give an aziridine molecule (molecular volumes of dinitrene biphenyl and dinitrene disulfonyl biphenyl are 0.1814 and 0.2132 nm3, respectively; in addition the nitrogen atom of sulfonylnitrene is situated out of an aromatic ring plane).
The results obtained are in good agreement with relative reactivities of arylnitrenes and sulfonylnitrenes in the crosslinking reactions of various polymers. The ratios of DM values for crosslinking with arylnitrenes and with sulfonylnitrenes (these ratios are shown in parentheses) are more than 1.0 in the systems with high mobilities of structural segments (i.e. low glass transition temperatures, Tg): in acrylate-siloxane copolymer (8.0), butadiene-acrylnitrile copolymer (2.9), triple butadiene-acrylnitrile-methacrylnitrile copolymer (16.0), and less than 1.0 in the systems with low structural mobility: in polyurethane (0.36), in polyvinylpirolidone (0.67), cyclized polyisoprene (0.19).
Table 1 shows that DM is increasing along the raw D2>D1>D3. The higher DM for the D1 against D2 one can be explain by longer wavelength absorbency of D2 compared with D1. The higher DM for D3 compared with the one for D1 can be caused by two reasons: (1) D3 absorbs at shorter wavelength than D1; (2) a double bond is more easily accessible in the case of D1.
The polymer structure can also influence a nitrene reactivity. In the case of linear polymer 5 substitution of 7 mol. % of methyl groups to vinyl ones gives the reactivity increase of only 1.27 times, but going from 2 to 1 gives its 70 times rise. It should be noted that changing the liquid reactivity for the solid state one produces usually so called "smoothing" of the reactivity [14]. However we have not observed the said smoothing! One can explain that by closed packing of double bonds making them being close to each other in the case of oligomer 1. Owing to this we should propose not only a nitrene reaction with the double bonds but a polymerization of the adjacent vinyl groups by means of the specialized "chain propagating" initiated by a triplet nitrene and/or a radical product produced from a triplet nitrene-double bond interaction. The possible mechanism of the propagating can be shown by the following scheme.
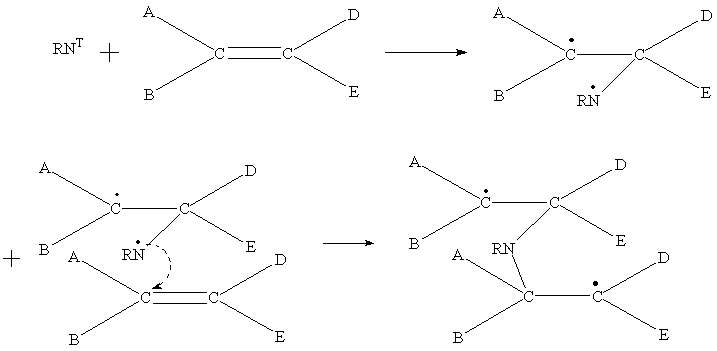 (2)
(2)
The reaction 2 might be relevant to explain the synergetic increase of the u.v. light sensitivity in solid films containing both cynnamate and azide groups [15].
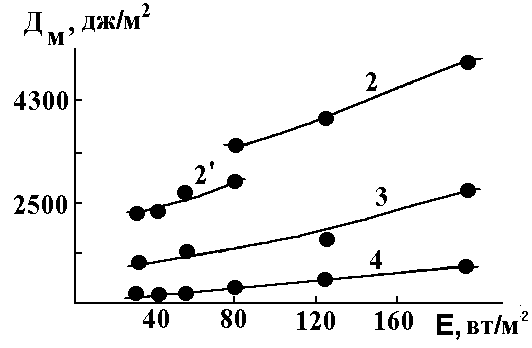
Figure 3. Changing of DM values under storing of the films made of the 1-D1 system. Here: figures above curves are time of storing in a day and night periods (24 h); 2’ – the film was coated an then stored 8 h before exposure.
We have also observed a different type of the curves DM for the linear and cyclolinear oligosiloxane films being irradiated with the u.v. light in presence of oxygen against the irradiation intensity, E (Fig.3). Indeed, for linear oligosiloxane and linear carboboned polymers DM increases, when E decreases (curve 2). But for system 1-D1 one can observe the curve having maximum. Both these curves differ tremendously from system containing cyclized polyisoprene-diazide 1 (see dotted line on the fig.3). The difference could be caused by some reasons: (1) The nitrene reactivity with a double bond is higher than with an ordinary (usually C-H) bond. The latter leads to decrease of the photooxidation contribution; and so as the photooxidation competes with the photocrosslinking, the latter leads to higher DM. (2) Oligomer 1 and diazide D1 form more close packing causing decrease in diffusion coefficient of O2. This leads to a lesser contribution of the photooxidation reaction. (3) Close neighborhood of azide and vinyl groups gives an increase of the crosslinking reactivity and diminishes the photooxidation role.
4. Conclusion.
In contrast to E-beam exposure of polysiloxanes where the crosslinking mechanism is HO-groups dehydration the u.v. light irradiation of a vinyl-containing oligosiloxane organic diazide compositions seems to take a course of an intermolecular aziridine linkage formation as a result of a nitrene attack to vinyl groups of the polymer. In the course of this study it was revealed that the reactivity of nitrenes in the compositions are different in the case of high and low reagent mobilities. In the former systems the relative reactivity of sulfonylnitrene is more than arylnitrene, their ratio being the same as one could predict on the basis of electronic structure. In the latter systems the relative reactivity ratio is reversed, the reactivity controlling factors being properties of the geometrical structure of the components of the reaction system such as the nitrene size, the interposition of vinyl groups of polymers, an ability for a nitrene molecule to come close to a double bond of a polymer.
It is strange enough not to observe the reactivity smoothing when one comes from reactivities of reagents in a reaction occurring in the liquid state to the reactivities of the same reagents but in the reaction having place in the solid state. The phenomenon was explained in terms of an additional polymerization giving a contribution to the crosslinking process. It seems us to be tempting to study conditions when that additional process gives the most valuable contribution. A very small contribution of the organic azide photooxidation in polysiloxane-containing systems is quite usual, too. We can not explain the phenomenon just nowadays.
Acknowledgment. The work was done under financial support of the INKAS organization (project 99-3-05) and Scientific Program "Russian University - Basic Researches" (project 015.05.01.38).
Reference