| Site home page | Conference home page | Discussion |
Quantum-chemical studies of the organic azide photooxidation mechanism
Sergei V. Zelentsov, Alexander A. Shchepalov
Nizhnii Novgorod State University
Gagarin Ave., 23. Nizhnii Novgorod 603600 Russia
E-mail: zelen@ichem.unn.runnet.ru
1. Introduction.
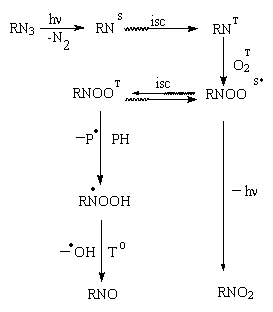
Recently it has been proposed a general reaction scheme of the organic azide photooxidation process [1]. The main idea of the scheme is a participation of the nitrene-oxygen adducts both in the triplet and singlet state. Their formation was found by Brinen and Singh [2]. The authors observed formation of both paramagnetic (having the ESR spectra typical for triplet biradical particles) and diamagnetic (yellowish) substances when they irradiated solutions of the azides in organic solvents saturated with O2 and frozen at 77 K. The main reactions on the scheme proposed [1] can be shown as following.
(Here S and T denote the singlet and triplet states, correspondingly).
The singlet nitrene generated under the u.v. light irradiation undergoes the intersystem crossing to give the triplet nitrene (The direct interaction of RNS in presence of molecular oxygen does not produce any oxygen-containing product [3]). The triplet nitrene reacts with oxygen to form the singlet nitrene-oxygen adduct in the (vibronically?) excited state. There are two possible routes to obtain stable products from the singlet adduct. The first one suggests the intersystem crossing into the triplet state to produce the triplet adduct. One of the possible reaction mechanism to stabilize the triplet adduct is its reaction with hydrogen-containing molecules of the reaction system. The final product from the triplet route is nitroso compound. The second route for the singlet adduct includes formation of the excited nitro compound molecules emitting an hemiluminescense quanta (The hemiluminescense was recently observed in [4,5]).
Unfortunately, there is scarce information about properties of the intermediates involved in the both routes. The intriguing question is the multiplicity of the adduct ground state. Some works say it is to be the triplet [6], other ones suggest it is to be the singlet state [7]. The other questions are concerned with the detailed mechanisms of the reaction routes and properties of the intermediates involved in..
We believe that some of the problems mentioned could be solved by means of modern quantum chemical calculations. The description of the results of such calculations and their discussion are aims of the present paper.
2. Methods of the quantum chemical calculations.
We have used ab initio methods such as UHF [8,9], ROHF [10,11], the CI family methods such as CASSCF [12,13], the Moller-Plesset perturbation theory of the second order (MP2) [14-16], the density functional theories (DFT) [17-19] using B3LYP functional [20], and semiempirical ones such as MNDO [21,22], AM1 [23] and PM3 [24,25]. Here slash separates the basis used; figures denote ways of representing of AOs as the gauss primitives expansions; the first characters in parentheses denote types of polarizing AOs added on the second period elements; AOs added on hydrogen atoms are shown after comma; and sign “++” denotes an addition of the diffusive functions on all atoms. In the case of CASSCF methods the figures in parentheses written just after word “CASSCF” are numbers of electrons and orbitals included into CI procedure (into active space).
The calculations were performed using the program PC GAMESS [26] and the program product Gaussian 94 [27]. The calculations were done at the Super Computer Center of the N.D. Zeleinsky Institute of Organic Chemistry of Russian Academy of Science.
3. Results and Discussion
3.1. The ground state multiplicity of the nitrene-oxygen adducts
As it was mentioned in Introduction there is scarce information concerning the nitrene-oxygen adduct properties, and especially about the multiplicity of the ground state of the adduct. It has been felt that the first step has to be brief investigation of the problem. Two systems, HNOO and PhNOO, were used as model the system being studied with CASSCF/6-31G (d) methods taking into account up to 10 electrons and 10 orbitals. It was found out that the most suitable was to use active space consisting of 8 electrons and 5 orbitals. Such wave functions were used to optimize geometrical parameters of HNOO both in the singlet and triplet state. The most important results are following.
Considerable size of the PhNOO system did not allow to perform the calculations in the same extend as for the HNOO system. In this case the wave functions of both the singlet and triplet state molecules contained only 3 Slater determinants. In the case of the Z-tautomerical form of PhNOO it was established the existence of two local minima on the potential energy surfaces (PES) of the adduct in the triplet and singlet states (See Fig.1).
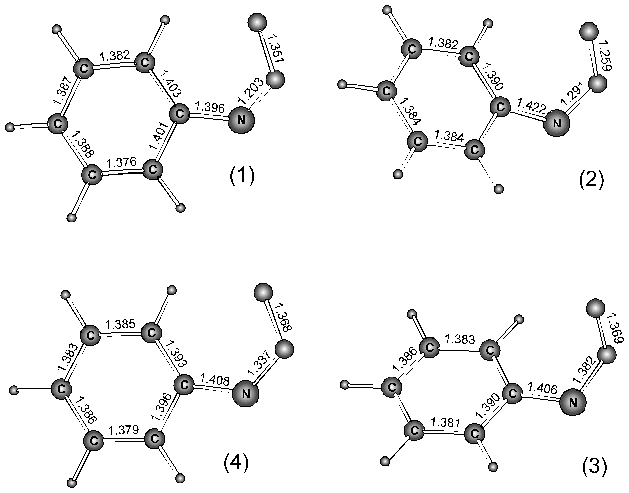
Fig.1. The local minima of the PhNOO potential energy surface: 1 and 2 are belonged to the singlet potential energy surface, and 3 and 4 to - the triplet one.
Adducts 1 and 2 are in the singlet, and 3 and 4 are in the triplet state. The adducts 1 and 4 have the planar geometries. In adduct 2 fragment CNOO is situated in the plane having angle with the phenyl ring plane of 47° . Nitrogen atom is slightly out of the phenyl ring plane. The local minimum corresponding to the adduct 2 geometry can not be found with the methods using one Slater determinant wave functions. This might be a consequence of a large biradical contribution into the wave functions. The adduct 3 has non-plane geometry too. In this case carbon bonded to nitrogen do not belong to the NOO plane, and nitrogen being situated in the phenyl ring plane.
For all stationary points found it has been calculated the energy of the state of other multiplicity, i.e. for the molecule being in the singlet state the triplet state molecule energy has been calculated and vice versa. To obtain more reliable results MP2 procedure has been applied to take into account the correlation corrections. 2 in its singlet state has the least energy. The energy of 3 in the triplet state is about 52 kJ M-1 greater than of 2. The singlet-triplet energy gap for 3 is only 2.5 kJ M-1. The latter value suggests that the probability of singlet-triplet transitions in the case of 3 might have considerable value. (See Fig.2).
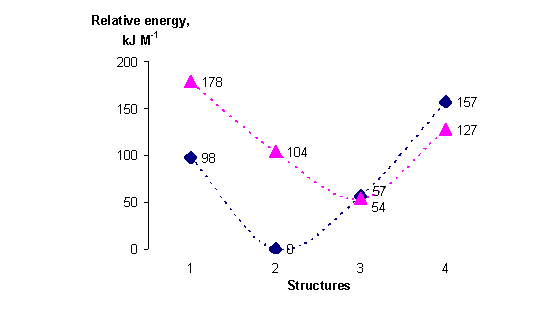
Fig.2. Relative energy gaps between the states of different multiplicity. Numbers of geometries of the adducts are defined on the Fig.1. (dark blue points denote the singlet PES and magenta ones – the triplet PES)
Thus the ground states of the nitrene-oxygen adducts seem to be siglet. This conclusion will be discussed later in this paper.
3.2. The triplet reaction route.
One of the main intermediates appearing in the triplet reaction route is N-radical hydroperoxide. The aim of this section is to investigate these unusual substances with modern quantum chemical methods.
The N-radical hydroperoxides as the organic azide photooxidation intermediates were proposed in [1,28,29]. They are formed as a result of an hydrogen atom abstraction molecules of the reaction media caused by the nitrene-oxygen adducts [28,29]
![]()
or as a result of trapping of hydroxyl radicals, •OH, by nitrosocompounds [30]
![]()
(Here R and R’ are hydrocarbonous substituents). The photooxidatiom scheme shown in Introduction says that a thermal dissociation of RN•OOH [29] is the main source of nitrocompounds
![]()
At present there are no reliable experimental data on the geometrical structure of the N-radical hydroperoxide RN•OOH, or on the energetic barrier of their dissociation. So we produced the quantum chemical calculations for RN•OOH where R=H, Me and Ph.
To verify the results obtained we compared the calculated and experimental geometrical parameters for •OH [30] and HNO [31] (See Tabl.1).
The data shown gave an opportunity to state that the best results were obtained with UB3LYP/6-31G(d,p), UMP2/6-311G(d,p), UHF/6-31G(d,p), and with AM1 when semiempirical methods were compared.
Table 1.
Comparison of the geometrical and thermodynamic data for HNO and •OH obtained with different theoretical methods and the known experimental data.
|
Method |
HNO |
•OH |
DfHo (g, 298.15K), kJ M-1 |
|||
|
H-N1) |
N-O1) |
H-N-O2) |
O-H1) |
HNO |
•OH |
|
| MNDO |
0.1048 |
0.1161 |
113.8 |
0.0937 |
14.73 |
2.13 |
| AM1 |
0.1042 |
0.1157 |
115.4 |
0.0949 |
9.12 |
3.84 |
| PM3 |
0.0997 |
0.1175 |
116.1 |
0.0937 |
57.78 |
11.89 |
| UHF/3-21G |
0.1036 |
0.1217 |
109.5 |
0.0986 |
- |
- |
| UHF/6-31G(d,p) |
0.1033 |
0.1175 |
108.9 |
0.0955 |
- |
- |
| UHF/6-311G(d,p) |
0.1032 |
0.1168 |
109.2 |
0.0952 |
- |
- |
| UHF/6-311++G(3df,2p) |
0.1031 |
0.1165 |
109.4 |
0.0950 |
- |
- |
| ROHF/6-31G(d,p) |
0.1033 |
0.1175 |
108.9 |
0.0954 |
- |
- |
| ROHF/6-311G(d,p) |
0.1032 |
0.1168 |
109.2 |
0.0951 |
- |
- |
| ROHF/6-311++G(3df,2p) |
0.1031 |
0.1165 |
109.4 |
0.0950 |
- |
- |
| UMP2/6-31G(d,p) |
0.1053 |
0.1237 |
107.2 |
0.0972 |
- |
- |
| UMP2/6-311G(d,p) |
0.1053 |
0.1222 |
107.6 |
0.0967 |
- |
- |
| UB3LYP/6-311G(d,p) |
0.1066 |
0.1200 |
108.6 |
0.0975 |
- |
- |
| G2 |
0.1058 |
0.1236 |
107.4 |
0.0979 |
97.10 |
35.11 |
| Experiment |
0.1063[31] |
0.1211[31] |
108.6[31] |
0.0970[30] |
102.03[32] |
39.34[32] |
1) – distance, nm; 2) – angle, degree.
The geometrical parameters of the N - radical hydroperoxides, RN•OOH, where R = H, Me, and Ph, obtained in our calculations are shown in Table 2-4. It should be noted that geometries of NOOH fragment calculated with different ab initio and semiempirical methods differ not greatly.
Table 2.
Geometries and energies of the HN•OOH system (HN•OOH, complex HNO + •OH and the transitional state of the HN•OOH dissociation reaction)
|
Method(basis) |
R(H-O)1) |
R(O-O) |
R(N-O) |
H-O-O2) |
O-O-N |
H-O-O-N |
total energy, a.u. |
D fHo(298), kJ M-1 |
|
Geometries of the HOON- fragment in radical HOON•H |
||||||||
| MNDO | 0.0965 | 0.1297 | 0.1286 | 109.4 | 111.8 | 100.7 | - | 71.80 |
| AM1 | 0.0984 | 0.1306 | 0.1292 | 108.7 | 112.0 | 83.7 | - | 143.72 |
| PM3 | 0.0942 | 0.1548 | 0.1279 | 95.7 | 100.7 | 179.9 | - | 107.86 |
| UHF/3-21G | 0.0973 | 0.1455 | 0.1437 | 101.3 | 105.8 | 94.8 | -204.01167 | - |
| UHF/6-31G(d,p) | 0.0948 | 0.1380 | 0.1334 | 103.3 | 108.5 | 83.8 | -205.14824 | - |
| UHF/6-311G(d,p) | 0.0945 | 0.1369 | 0.1330 | 103.6 | 109.0 | 84.7 | -205.20513 | - |
| UHF/6-311++G(3df,2p) | 0.0944 | 0.1368 | 0.1325 | 103.9 | 108.9 | 84.4 | -205.22991 | - |
| ROHF/6-31G(d,p) | 0.0948 | 0.1380 | 0.1336 | 103.3 | 108.4 | 84.7 | -205.14262 | - |
| ROHF/6-311G(d,p) | 0.0945 | 0.1368 | 0.1331 | 103.7 | 108.9 | 83.7 | -205.19946 | - |
| ROHF/6-311++G(3df,2p) | 0.0944 | 0.1368 | 0.1327 | 104.0 | 108.8 | 84.8 | -205.22340 | - |
| CASSCF(5,5)/6-31G(d,p) | 0.0949 | 0.1473 | 0.1390 | 100.1 | 105.7 | 90.2 | -205.21865 | - |
| CASSCF(5,5)/6-311G(d,p) | 0.0946 | 0.1465 | 0.1329 | 100.3 | 107.2 | 96.1 | -205.26095 | - |
| CASSCF(5,5)/6-311++G(3df,2p) | 0.0945 | 0.1460 | 0.1325 | 100.9 | 107.2 | 91.8 | -205.28444 | - |
| CASSCF(9,9)/6-311G(d,p) | 0.0970 | 0.1448 | 0.1377 | 100.4 | 107.0 | 82.3 | -205.34561 | - |
| CASSCF(9,9)/6-311++G(3df,2p) | 0.0945 | 0.1453 | 0.1372 | 101.1 | 106.8 | 84.1 | -205.36757 | - |
| UMP2/6-31G(d,p) | 0.0973 | 0.1449 | 0.1350 | 100.0 | 107.1 | 59.0 | -205.66145 | - |
| UMP2/6-311G(d,p) | 0.0967 | 0.1431 | 0.1336 | 100.3 | 107.9 | 60.7 | -205.75811 | - |
| UB3LYP/6-31G(d,p) | 0.0979 | 0.1446 | 0.1341 | 99.9 | 108.1 | 0.0 | -206.21155 | - |
| UB3LYP/6-311G(d,p) | 0.0975 | 0.1443 | 0.1335 | 100.2 | 108.3 | 0.0 | -206.27106 | - |
| G2 | 0.0981 | 0.1449 | 0.1350 | 100.1 | 107.2 | 56.6 | -205.94963 | 151.58 |
|
Geometries of the HOON- fragment in the HO+HNO complex |
||||||||
| MNDO | 0.0937 | 0.4561 | 0.1161 | 18.2 | 96.4 | 179.5 | - | 3.73 |
| AM1 | 0.0950 | 0.3169 | 0.1158 | 5.4 | 117.2 | 179.3 | - | 1.22 |
| PM3 | 0.0939 | 0.1955 | 0.1178 | 103.1 | 116.2 | 180.0 | - | 16.11 |
| PM31) | 0.0938 | 0.3549 | 0.1185 | 0.3 | 151.1 | 6.2 | - | 23.18 |
| UHF/3-21G | 0.0984 | 0.3054 | 0.1353 | 3.4 | 171.4 | 0.0 | -204.03280 | - |
| UHF/6-31G(d,p) | 0.0956 | 0.3143 | 0.1177 | 1.3 | 111.6 | 178.6 | -205.18279 | - |
| UHF/6-311G(d,p) | 0.0953 | 0.3170 | 0.1191 | 1.8 | 118.6 | 98.7 | -205.24302 | - |
| UHF/6-311++G(3df,2p) | 0.0952 | 0.3264 | 0.1212 | 9.9 | 119.6 | 116.3 | -205.26593 | - |
| ROHF/6-31G(d,p) | 0.0956 | 0.3139 | 0.1177 | 1.5 | 111.5 | 170.3 | -205.17896 | - |
| ROHF/6-311G(d,p) | 0.0953 | 0.3145 | 0.1169 | 1.6 | 116.1 | 178.7 | -205.23671 | - |
| ROHF/6-311++G(3df,2p) | 0.0952 | 0.3166 | 0.1167 | 0.3 | 115.3 | 166.6 | -205.26044 | - |
| CASSCF(5,5)/6-31G(d,p) | 0.0956 | 0.3155 | 0.1232 | 0.3 | 107.4 | 68.8 | -205.27201 | - |
| CASSCF(5,5)/6-311G(d,p) | 0.0952 | 0.3156 | 0.1225 | 0.3 | 114.6 | 9.8 | -205.32945 | - |
| CASSCF(5,5)/6-311++G(3df,2p) | 0.0973 | 0.3269 | 0.1196 | 1.1 | 112.6 | 4.4 | -205.32805 | - |
| CASSCF(9,9)/6-311G(d,p) | 0.0976 | 0.3237 | 0.1225 | 0.6 | 113.6 | 83.3 | -205.37798 | - |
| CASSCF(9,9)/6-311++G(3df,2p) | 0.0952 | 0.3204 | 0.1216 | 2.5 | 112.2 | 1.9 | -205.41042 | - |
| UMP2/6-311G(d,p) | 0.0969 | 0.3068 | 0.1224 | 0.1 | 109.8 | 178.7 | -205.77412 | - |
| B3LYP/6-31G(d,p) | 0.0976 | 0.2356 | 0.1209 | 83.2 | 110.6 | 106.1 | -206.20422 | - |
| B3LYP/6-31G(d,p)2) | 0.0983 | 0.3038 | 0.1211 | 0 | 110.9 | 1.6 | -206.20613 | - |
| B3LYP/6-311G(d,p) | 0.0972 | 0.2348 | 0.1201 | 83.4 | 111.2 | 115.6 | -206.26851 | - |
| B3LYP/6-311G(d,p)3) | 0.0979 | 0.3030 | 0.1203 | 0.0 | 115.0 | 177.5 | -206.27023 | - |
|
Geometries of the HOON- fragment in the transitional state of the reaction HNOOH -> HNO+OH |
||||||||
| PM3 | 0.0939 | 0.1678 | 0.1239 | 94.0 | 102.9 | 179.9 | - | 27.48 |
| UHF/3-21G | 0.0976 | 0.1537 | 0.1420 | 97.3 | 103.7 | 177.1 | -204.00936 | - |
| UHF/6-31G(d,p) | 0.0950 | 0.1565 | 0.1315 | 98.0 | 106.7 | 103.9 | -205.13801 | - |
| UHF/6-311G(d,p) | 0.0947 | 0.1555 | 0.1307 | 97.3 | 105.9 | 173.1 | -205.19313 | - |
| CASSCF(5,5)/6-31G(d,p) | 0.0952 | 0.1700 | 0.1373 | 94.0 | 102.0 | 179.5 | -205.20870 | - |
| UB3LYP/6-311G(d,p) | 0.0970 | 0.1693 | 0.1247 | 95.8 | 109.3 | -150.7 | -206.26021 | - |
1) – distance, nm; 2) – angle, degree.
Table 3.
Geometries and energies of the CH3N•OOH system (CH3N•OOH, complex CH3NO + •OH and the transitional state of the CH3N•OOH dissociation reaction).
|
Method(basis) |
R(H-O)1) |
R(O-O) |
R(N-O) |
H-O-O2) |
O-O-N |
H-O-O-N |
total energy, a.u. |
D fHo(298), kJ M-1 |
|||
|
Geometries of the HOON- fragment in radical CH3N•OOH |
|||||||||||
| AM1 |
0.0984 |
0.1302 |
0.1306 |
108.7 |
111.9 |
83.9 |
- |
32.05 |
|||
| PM3 |
0.0944 |
0.1515 |
0.1310 |
96.1 |
99.4 |
179.8 |
- |
21.78 |
|||
| UHF/6-31G(d,p) |
0.0948 |
0.1380 |
0.1343 |
103.4 |
108.5 |
81.3 |
-244.18846 |
- |
|||
|
Geometries of the HOON- fragment in the transitional state of the reaction CH3NOOH -> CH3NO+OH |
|||||||||||
| PM3 |
0.0939 |
0.1703 |
0.1245 |
93.6 |
102.5 |
179.8 |
- |
26.08 |
|||
|
Geometries of the HOON- fragment in the HO+ CH3NO complex |
|||||||||||
| AM1 |
0.0949 |
0.2333 |
0.116 |
72.7 |
117.1 |
179.7 |
- |
-4.54 |
|||
| PM3 |
0.0938 |
0.1954 |
0.1182 |
105.5 |
117.7 |
119.8 |
- |
9.22 |
|||
1) – distance, nm; 2) – angle, degree.
Table 4.
Geometries and energies of the PhN•OOH system (PhN•OOH, complex PhNO + •OH and the transitional state of the PhN•OOH dissociation reaction).
|
Method(basis) |
R(H-O)1) |
R(O-O) |
R(N-O) |
H-O-O2) |
O-O-N |
H-O-O-N |
total energy, a.u. |
D fHo(298), kJ M-1 |
|
Geometries of the HOON- fragment in radical PhN•OOH |
||||||||
| AM1 |
0.0985 |
0.1298 |
0.1318 |
108.8 |
111.0 |
83.7 |
- |
59.02 |
| PM3 |
0.0944 |
0.1515 |
0.1318 |
96.1 |
98.6 |
179.1 |
- |
49.40 |
| UHF/6-31G(d,p) |
0.0948 |
0.1377 |
0.1347 |
103.4 |
107.9 |
81.0 |
-434.72773 |
- |
| MP2/6-31G(d,p) |
0.0973 |
0.1453 |
0.1371 |
99.7 |
106.3 |
66.7 |
-436.00354 |
- |
| B3LYP/6-31G(d,p) |
0.0978 |
0.1446 |
0.1355 |
99.7 |
107.2 |
0 |
-437.28494 |
- |
|
Geometries of the HOON- fragment in the transitional state of the reaction PhNOOH -> PhNO+OH |
||||||||
| PM3 |
0.0940 |
0.1656 |
0.1259 |
94.1 |
102.6 |
149.8 |
- |
52.84 |
|
Geometries of the HOON- fragment in the HO+ PhNO complex |
||||||||
| AM1 |
0.0950 |
0.2890 |
0.1161 |
37.8 |
113.6 |
118.9 |
- |
29.01 |
| PM3 (complex 1) |
0.0938 |
0.1953 |
0.1183 |
105.4 |
117.9 |
114.8 |
- |
41.42 |
| PM3 (complex 2) |
0.0938 |
0.3523 |
0.1193 |
8.7 |
141.8 |
145.7 |
- |
49.00 |
| UHF/6-31G(d,p) |
0.0956 |
0.3162 |
0.1221 |
4.6 |
117.0 |
122.1 |
-434.76102 |
- |
Almost all methods used gave a non-planar structure of HN•OOH. Results of the UHF/6-311G(d,p) method (which were typical) ones were the following: R(H-O) = 0.0945 nm, R(O-O) = 0.1369 nm, R(O-N) = 0.1330 nm, Ð (H-O-O) = 103.6° , Ð (O-O-N) = 109.0° , Ð (H-O-O-N) = 84.7° . The most noticeable difference of results was observed when the 3-21G basis was replaced with the 6-31G(d,p) one. It is evident that this difference is caused by addition of the diffusive orbitals on hydrogen and second period atoms. Further expanding of the basis gives only 0.001 nm shortening of O-O and N-O bonds and 0.0004 nm shortening of O-H bond. Substitution of ROHF for UHF method has no influence on the calculated geometrical parameters. CASSCF use gives elongation of O-O bond for 0.08-0.09 nm and O-N bond for 0.002-0.005 nm compared to the results obtained with the UHF method. Bond lengths obtained with CASSCF, MP2 and DFT B3LYP procedures are differing within less than 0.001 nm interval.
Substitution of hydrogen for methyl and phenyl group gives no substantial changing of the results.
DFT B3LYP and PM3 give different geometrical parameters of the N-radical hydroperoxide. In this case all atoms of NOOH fragment is situated on the same plane, the NOOH fragment structures can be represented by
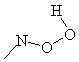
![]()
5 6
the structure 6 having NOOH angle of 180° and being more stable due to MP3 method and structure 5 having NOOH angle 0° and being more stable according to results obtained with the DFT B3LYP method.
The distinction between the results given by B3LYP and the results of other methods seems to be explained with overestimation of the electron correlation contribution that leads to geometry distortions and an additional stabilization (over-stabilizing effect) of the RN•OOH radical and to considerable total energy decreasing.
Fig.3 shows electron density distribution in the RN•OOH radicals. Almost all spin density of HN•OOH radical are localized on the nitrogen atom (more than 90%) and partly on the oxygen atom adjacent to the nitrogen atom (structures 7 and 8). There is no qualitative distinction of the spin density distributions obtained by the one-determinant (structure 7) and the multi-determinant methods (structure 8). So we can conclude that it is enough to use only the one-determinant methods to find out estimations of radical geometrical parameters and spin density distribution.
Substitution of hydrogen atom for methyl group gives only insignificant changing of spin density distribution (see structure 9 on Fig.3). Substitution of hydrogen atom for phenyl group produces partial redistribution of spin density, namely a shift of it into the phenyl ring due to conjugation of the NOO fragment with the aromatic system causing formation of the united p -system (structure 10 on Fig.3).
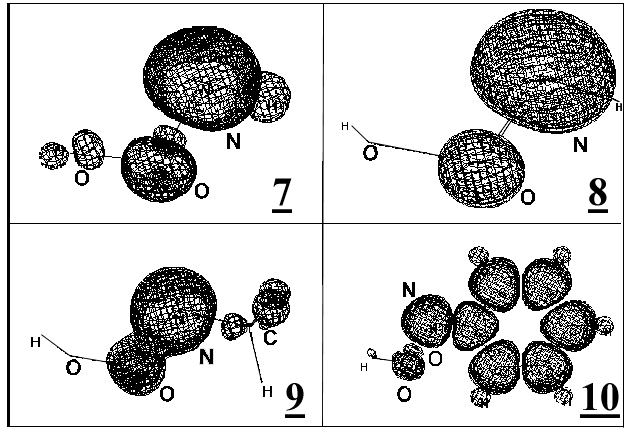
Fig.3. Spin density distribution in the RN•OOH radical:
7 – radical HN•OOH, UHF/6-31G(d,p) calculation; 8 – radical HN•OOH, CASSCF (9,9)/6-311G (d,p) calculation; 9 – radical MeN•OOH, UHF/6-31G(d,p) calculation; 10 - radical PhN•OOH, UHF/6-31G(d,p) calculation.
Fig.4 shows the energy profile of the HN•OOH dissociation reaction (the reaction coordinate is length of O-O bond) calculated by means of UHF/6-31G(d,p) method.
When the length of O-O bond is 0.156 nm the transitional state will have place. Its geometrical parameters are R(H-O) = 0.095 nm, R(O-O) = 0.1565 nm, R(O-N) = 0.1315 nm, Ð (H-O-O) = 98° , Ð (O-O-N) = 106.7° , Ð (H-O-O-N) = 103.9° . Energy barrier of the reaction is 26.86 kJ M-1. Geometrical parameters of the radicals calculated with different methods are shown in the lower parts of the Tables 2-4.
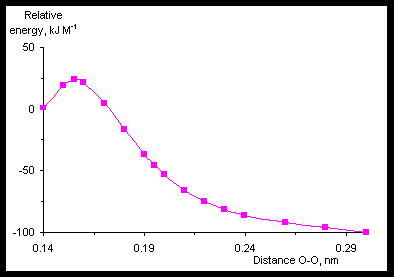
Fig.4. Energy profile of the model radical HN•OOH dissociation reaction (calculated with UHF/6-31G(d,p) method).
The CASSCF(5,5)/6-31G(d,p) calculation did not give “a real transitional state”. Cross-section of the potential energy surface of the HN•OOH was built. To do this the length of the O-O bond was fixed, and the remained geometrical parameters were optimized, i.e. it was used so-called “relaxed scan”. There was a maximum at the length of the O-O bond from interval 0.165-0.170 nm on the cross-section. Energy of “the transitional state” was 26.11 kJ M-1 that is not far from 26.86 kJ M-1 obtained with UHF/6-31G(d,p).
The DFT B3LYP/6-311G(d,p) calculations were also used to find the geometrical parameters of the transitional state for the HN•OOH decomposition under the O-O bond elongation. The found transitional state had the only imaginary vibrational mode of 527.7 cm-1, the O-O bond length increase having been of 0.026 nm, and the N-O bond contraction having been of 0.0092 nm with respect to the optimized geometrical parameters of HN•OOH. Dihedral angle H-O-O-N becomes by 30° wider. Energy barrier is of 28.49 kJ M-1.
Thus we can conclude that when the system moves along the reaction coordinate the main geometrical changes are (1) enlargement of the dihedral angle N-O-O-H by 20-30° , and (b) elongation of the O-O bond by 0.02 nm, the latter means that the geometric structure of the starting peroxide is very close to the one of the transitional state.
The spin density located on atoms changed in the course of the reaction. It is clearly seen from Fig.4 that electrons unpairedness of the O-O bond in the transitional state is of only 35%. Further enlargement of the O-O distance leads to a noticeable shift of spin density to hydroxyl oxygen (O2). Spin density on the nitrogen atom diminishes too. It is should be noted that spin densities on the nitrogen atom and on the middle oxygen atom were decreasing synchronously. The latter supports the idea of the N-O bond formation.
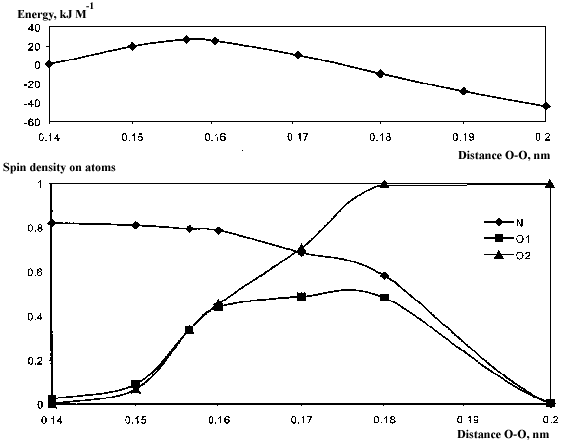
Fig.5. HN•OOH radical dissociation (calculated with the UNF/6-31G (d,p) method): 1 –the energy profile of the reaction; 2 – spin density variation on the atoms from N-01-02 fragment.
Let us discuss the intermediates which formation occurred at further movement along the reaction coordinate of the HN•OOH radical destruction (See the middle part of Table 2-4).
When the B3LYP/6-311G(d,p) or PM3 method having been used, the primary product was complex 11 where hydroxyl was coordinated with nitrosogroup due to the unpaired electron
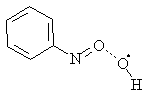
11
Geometrical parameters of the complex 11 (HNO + •OH system, B3LYP/6-311G (d, p) calculation) were R(H-O) = 0.0972 nm, R(O-O) = 0.2348 nm, R(O-N) = 0.1201 nm, Ð (H-O-O) = 83.4 ° , Ð (O-O-N) = 111.2° , Ð (H-O-O-N) = 115.6° . Heat of the reaction was 6.70 kJ M-1. The obtained geometry corresponded to the local minimum. The latter conclusion based upon the fact that an analysis of the vibration spectrum did not reveal any imaginary vibration. Almost all spin density was located on the hydroxyl oxygen (94%) and only part it was on the oxygen atom of nitroso group.
This complex was not found with other methods including the MP2 procedure but only with PM3 and B3LYP/6-311G(d,p) methods.
Further movement along the reaction coordinate gave the complex 12 arising from the hydroxyl radical coordination onto the oxygen atom of the nitroso group and hydrogen bond formation
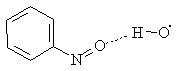
12
The complex 12 formation was found with all methods used. Here the distance between oxygen atoms was more than 0.3 nm, and length of the hydrogen bond formed was 0.20 - 0.22 nm (varied with the method used).
In the case of the PhN•OOH radical there was observed new complex formation arising from OH group coordination with the oxygen atom of nitroso group and H-atom of the phenyl ring (complex 13)
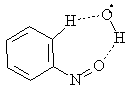
13
Table 5.
Heat of reaction (3) calculated by different methods
|
Method |
Heat of reaction, kJ M-1 |
| UHF/6-31G(d,p) |
-76.6 |
| UHF/6-311G(d) |
-78.7 |
| UHF/6-311G(d,p) |
-79.5 |
| UHF/6-311G(2d,p) |
-77.0 |
| UHF/6-311G(2d,2p) |
-76.6 |
| UHF/6-311G(3df,2p) |
-75.3 |
| UHF/cc-pVTZ |
-78.7 |
| UHF/cc-pVTZ |
-79.1 |
| UHF/aug-cc-pVDZ |
-84.1 |
| UHF/6-311++G(d,p) |
-86.2 |
| UHF/6-311++G(2d,p) |
-84.5 |
| UHF/6-311G++(2d,2p) |
-84.1 |
| UHF/6-311++G(3df,2p) |
-82.0 |
| UHF/aug-cc-pVTZ |
-81.2 |
| ROHF/6-31G(d,p) |
-81.2 |
| ROHF/6-311G(d,p) |
-84.1 |
| ROHF/6-311++G(3df,2p) |
-86.6 |
| UMP2/6-31G(d,p) |
-15.1 |
| UMP2/6-311G(d) |
-25.9 |
| UMP2/6-311G(d,p) |
-25.5 |
| UMP2/6-311G(2d,p) |
-10.5 |
| UMP2/6-311G(2d,2p) |
-10.5 |
| UMP2/6-311G(3d,2p) |
-10.5 |
| UMP2/6-311G(3df,2p) |
-0.4 |
| UMP2/cc-pVTZ |
-3.3 |
| UMP2/6-311++G(d,p) |
-32.6 |
| UMP2/6-311G++(2d,p) |
-19.2 |
| UMP2/6-311++G(2d,2p) |
-18.8 |
| UMP2/6-311++G(3df,2p) |
-10.9 |
| UMP2/aug-cc-pVTZ |
-5.9 |
| UB3LYP/6-31G(d,p) |
32.6 |
| UB3LYP/6-311G(d) |
19.7 |
| UB3LYP/6-311G(d,p) |
20.1 |
| UB3LYP/6-311G(2d,p) |
27.6 |
| UB3LYP/6-311G(2d,2p) |
27.6 |
| UB3LYP/6-311G(3df,2p) |
28.0 |
| UB3LYP/cc-pVTZ |
23.0 |
| UB3LYP/6-311++G(d,p) |
5.4 |
| UB3LYP/6-311++G(2d,p) |
13.8 |
| UB3LYP/6-311++G(2d,2p) |
13.8 |
| UB3LYP/6-311++G(3df,2p) |
14.2 |
| MP4(SDQ)/6-31G(d,p) |
-23.8 |
| MP4(SDQ)/6-311G(d,p) |
-34.7 |
| MP4(SDTQ)/6-31G(d,p) |
-20.9 |
| MP4(SDTQ)/6-311G(d,p) |
-29.7 |
| QCISD/6-31G(d,p) |
-15.1 |
| QCISD/6-311G(d,p) |
-25.9 |
| CCD/6-31G(d,p) |
-28.5 |
| CCD/6-311G(d,p) |
-37.2 |
| CCSD/6-31G(d,p) |
-18.4 |
| G2 |
-21.8 |
| G2MP2 |
-20.5 |
Table 5 summarizes energetic characteristics of the reaction. It is easy to notice from the data shown that there is quite considerable variation among values obtained with different methods. Two groups of the methods could be found: (a) the methods taking the dynamic electron correlation into account (MP2, G2, etc.) and (b) methods without corrections caused by dynamic electron correlation (UHF). The former group gives little heat of the reaction, the heat being 15–25 kJ M-1 for the HN•OOH radical dissociation. The latter group of the methods performs more noticeable effect (of some times greater). It should be noted that great influence of the electron correlation on the heats of reaction was mentioned before [33-36].
Thus, we can conclude that the N-hydroperoxide radical decomposition is exothermal and has heat of about 22 kJ M-1.
As well as the abstraction of an hydrogen atom by the terminal oxygen at the triplet nitrene-oxygen adduct [29] there exists possibility of the abstraction by a nitrogen atom of the adduct. This possibility was pointed out in [28]. Some methods were used to investigate such a possibility of and to estimate its involvement in the photooxidation process. Relative stabilities of the intermediates and the possibility of their interconversion were studied. The results obtained are shown on Table 7.
Table 7.
Comparison of N-hydroperoxide radical and aminoperoxy radical
|
Method |
Energy difference, kJ M-1 |
|
between H2NOO and HNOOH |
|
| PM3 |
40 |
| B3LYP/6-31G(d) |
-25.7 |
| QCISD/6-311+G(2d) |
-16.7 |
| G2 |
-3.3 |
|
between C6H5N(H)OO and C6H5NOOH |
|
| B3LYP/6-31G(d) |
16.7 |
As one can see the PM3 gives very difference results from ab initio and DFT methods. The PM3 method parameterization procedure did not include molecules such as our radicals, so this method can not give good results for this class of molecules.
These radicals, H2NOO (14) and (15), have very similar energies and the relative stability depends of substitutes at nitrogen. By B3LYP/6-31G(d) method it was found that energy barrier for rearrangement reaction from H2NOO to HNOOH is 150.8 kJ M-1.
Using B3LYP/6-31G(d) method, the dissociation of amino peroxide radical 14 to give molecular oxygen and amino radical 17 was modeled. The result gives on Figure 7.
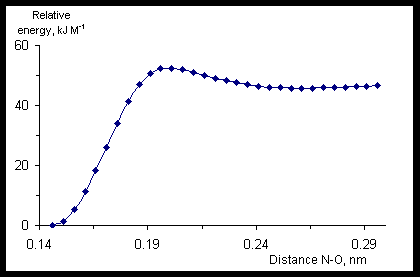
Figure 7. Decomposition of H2NOO• radical to amine radical and triplet oxygen.
“Rigid scan procedure”, i.e. when the O-N distance in the optimized geometry of H2NOO• was increasing, and the remainder geometrical parameters of the molecule were kept constant, was used. Energy barrier for this reaction was 52.3 kJ M-1, and heat of the reaction was 50.5 kJ M-1.
These values give an evidence of relatively small possibility of H2NOO participation in formation of the oxygen-containing photooxidation products.
3.3. The singlet reaction route intermediates.
The detailed description of the singlet state potential surface of RNO2 will be given elsewhere [39]. In addition, it should be noted that recently some excellent works on the theme have appeared [37,40,41]. The results of the works are summarized on Figure 8.
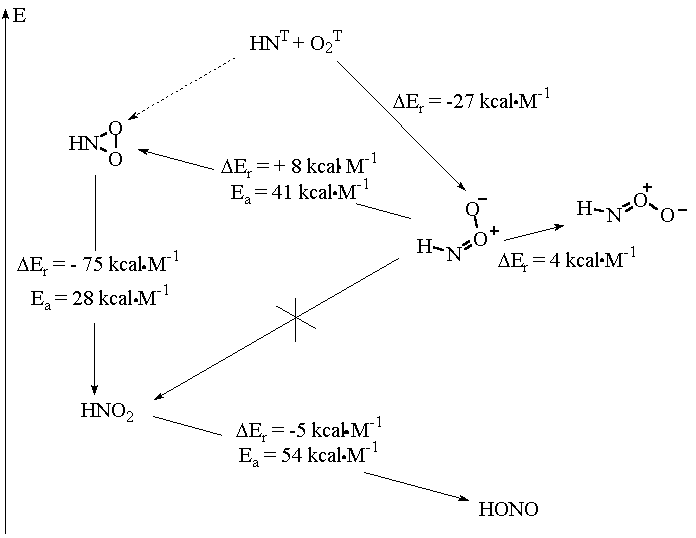
Figure 8. The Potential Energy Surface HNO2 calculated by B3LYP/6-31G(d). Ea – activation energy, DEr – heat of reaction.
Fueno and coworkers [40,41] have studied potential energy surface HNO2 using a multireference configuration interaction method. They have evaluated the rate constant of the reaction
![]()
Ngyen and coworkers [37] have studied this surface in more details. Owing to the results described in [37,39], we should note the following:
(a) the singlet adduct HNO2 can rearrange to nitro compound via intermediate formation of a cyclic singlet adduct [37,39]
![]()
(b) the rearrangement is highly exothermal, but the location of the linear RNOO, its cyclic form and nitro compound are separated by considerable energetic barriers of about 41.8 and 24.7 kcal M-1, respectively [37];
(c) the global minimum on the surface corresponds to nitrous acid, HONO; nitro compounds is situated only of 7.1 kcal higher, but the locations of the species are separated by very high barrier of 54.7 kcal M-1 [37,39];
(d) the main features of the HNO2 surface do not change in the case of PhNO2 surface [39].
This data will help us to discuss the photooxidation mechanism more appropriately.
4. Photochemical consequences of the results obtained.
One of the main results obtained in our paper was the fact that RNOOT and RNOOS had close energy, and RNOOS being the ground state particle. The latter conclusion is in a seeming contradiction with observation of the ESR spectra typical for biradicals. The contradiction might be eliminated only if RNOOS was generated with surplus energy. This has place because of energy emerging from triplet annihilation (O2T and the triplet nitrene). This surplus energy can make more probable intersystem crossing and be a cause of hemiluminescent quanta emitted by RNOOS* when it rearranges into nitro compound.
5. Acknowledgment.
The work was done under financial support Scientific Program "Russian University - Basic Researches" (015.05.01.38).
6. References