| Site home page | Conference home page | Discussion |
Structural pathways
of electron transfer via tunnelling and
van der Waals in the
cytochrome f-plastocyanin cpmplex
Mário Fragata
Université du Québec à Trois-Rivières,
Département de chimie-biologie,
Section de chimie, Trois-Rivières, Que, G9A 5H7, Canada
fragata@uqtr.uquebec.ca
TABLE OF CONTENTS
Abstract
1. Introduction
2. Methodology
3. Results and discussion
3.1.
Molecular structure of the docked interface in cyt f-pc
3.2.
Structural pathways of electron transfer in cyt f-pc
3.3.
Intramolecular electron transfer: Tunnelling effect
3.4.
Intramolecular electron transfer: Van der Waals contact
4.
Concluding remarks
5.
Abbreviations
References
Acknowledgements
Last page
A study was undertaken of the molecular pathways of electron transfer
in the cytochrome f-plastocyanin (cyt f-pc )
complex. The calculated centre-to-centre distances of electron
transfer (ETd) are (i) 7.2 Å from the Fe-atom and the tyrosine
1 (Y1) in cyt f, and (ii) 5.9 Å from Y1 to the Cu-atom in
pc. Within the plastocyanin core, ETd = 7.0 Å between
cysteine
84 (C84) and serine 85 (S85) and 5.2 Å between S85(OH) and tyrosine
83 [Y83(OH)]. A calculated alternative route of electron transfer
in pc is between C84(SH) and the p-electron
system in Y83 where ETd is ~7.3 Å. On the other hand,
the electron transfer distance between the Cu-atom and C84(SH) is approximately
2.6 Å, i.e., the coordination distance from the copper-atom to the
SH group in C84. ETd = 2.6 Å is clearly quite small in
relation to the 5 to ~7 Å determined for the various electron transfer
pathways in cyt f-pc. In this respect, it is noted first that electron
transfer distances higher than about 5 Å are in general a good indication
of electron transfer occurring by tunnelling efffect, that is, by a quantum
jump from one electronic state to another. On the contrary, it is
reasonable to assume that electron transfer reactions at distances as small
as 2.6 Å might simply take place via van der Waals contacts.
This is a convincing argument to suggest the coexistence of different molecular
mechanisms of electron transfer in the cyt f-pc complex. Such coexistence
of molecular states has far reaching consequences
so far as it presupposes the function in cyt f-pc of adiabatic and non-adiabatic
electron transfer processes which have different temperature dependence,
or requirements.
Biological electron transfer between redox proteins is
of fundamental importance in such speciallized organelles as the chloroplasts
of green plants and algae, or the mitochondria of most organisms.
In short, these molecular systems are responsible for the function and
the
efficiency of the photosynthetic and respiratory processes in living
cells. In nature, this question has been solved with a complex, yet
elegant system of molecular pathways leading to a remarkably efficient
transfer of electrons through molecular distances that can be about 20-25
Å, or longer (see data discussed in [1,2]). An interesting
aspect of this question is that the biological electron transfer devices
seem in general protected against difficulties brought about by energy
losses of various types. For example, electron transfer decay due
to inadequate molecular rearrangements in the structural pathways, or to
electron back reactions. This question has always been intriguing
since the construction of efficient man-made batteries for solar energy
conversion into chemical or electrical energy is quite often plagued with
difficulties inherent to energy losses due to the back reactions.
In this perspective, understanding at the molecular level the electron
transfer mechanisms as those taking place in the photosynthetic membrane
of the chloroplast might eventually foster our comprehension of how to
build artificial electron transfer devices with efficiencies comparable
to those usually observed in nature.
The study of the biological electron transfer mechanisms
has made the object of a very large number of works (see, e.g., [2-10]).
For the purposes of the present work, it is noted that in oxygenic photosynthesis
the cytochrome b6f (cyt b6f) complex transfers electrons
between photosystem II and the P700 reaction center of the photosystem
I complex [9]. From cytochrome f (cyt f) in cyt b6f to PSI,
the electrons are shuttled by the mobile redox carriers plastocyanin (pc)
and cytochrome c6 (cyt c6) [9,10]. In several cyanobacterial
species, cyt c6 is the only electron carrier but in other cyanobacteria
and in certain eukaryotic algae this role is played by either cyt c6 or
pc [3-7]. In those cases, the biosynthesis of either pc or
cyt c6 is determined only by the availability of copper in the environment
or in the culture medium. In higher plants, however, the genes for
cyt c6 are not present, meaning that pc is the sole electron carrier between
the cyt b6f complex and P700.
In spite of the many efforts to solve the electron transfer
question in the cyt f-pc system, the molecular mechanisms and structural
pathways of electron transfer in the photosynthetic membrane are yet far
from being clearly understood. The purpose of this study is twofold.
First,
the purpose is to model the most relevant structural pathways of electron
transfer in the cytochrome f-plastocyanin (cyt f-pc) with the intent of
devising a structure-function model electron transfer by tunnelling effect
or via van der Walls contacts. Secondly, the object of this
work is to use semiempirical mathematical expressions of electron tunnelling
to predict experimental rates of electron transfer, i.e., the electron
tunnelling probabilities. In this context, the present study showed
that the most efficient structural pathways of electron transfer in the
cyt f-pc complex are those characterized by small differences (V-E) between
the kinetic energy of the approaching electron (E) and the energy (V) of
the barrier above E, i.e., dV (=V-E, or the barrier height). That
is, dV is found to be approximately 0.1 eV at tunnelling distances between
5 and 7 Å.
In the determination of docking configurations and other molecular modellizations, we took into account the data obtained from chemical cross-linking, chemical modification of amino acid residues, and the effect of mutations on the reactivity of different pc and cyt f forms [11-17]. Special attention was also given to the structure of the interfacial medium between the heme in cyt f and the surfaces of docking of the electron acceptors (pc). As a rule, we took into account any structures in the surfaces of docking that may influence the donor-acceptor couplings (see discussion in [18]) in spite of the fact that some of those amino acid residues are not directly involved in the activity of the cyt f-pc complex. We have thus minimized the stereochemical hindrance at the interacting surfaces, but making sure at the same time that the molecular complementarity that is needed for efficient docking and electron transfer was preserved.
The atomic coordinates of cytochrome f and plastocyanin
were obtained from the Protein Data Bank [19,20], that is, 5pcy.pdb
(plastocyanin)
and
1ctm.pdb (cytochrome f). The cyt f-pc complexes
was modelled using the computer graphics program TURBO-FRODO
[21,22], and the structures were further refined with the X-PLOR
program (version 3.1) for the energy minimization (see details in [23,24]).
Some atomic distances and other molecular details were determined with
the WebLab ViewerPro software from Molecular Simulations Inc. (San Diego,
CA). Other calculations were performed with Maple (version V) from
Waterloo Maple Inc. (Waterloo, ON), and Origin (version 5) from Microcal
Software, Inc. (Northampton, MA).
3.1. Molecular structure of the docked interface in cyt f-pc
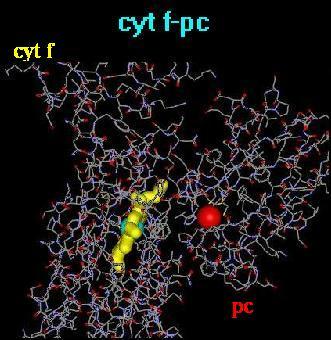
The structural representation of the cyt f and pc redox
partners in the cyt f-pc docked complex is displayed in Fig. 1.
This structure was obtained from a series of modellizations performed according
to the procedures which are described above (see Methodology). The
analysis of the structures represented in Fig. 1 [A. Kajava, M.
Fragata, in preparation] disclosed some predominant molecular details
in the docked surfaces of cyt f and pc such as the ionic bonds and charge
interactions on the one hand, and the hydrophobic contacts on the other
hand. The most important molecular details are summarized in the
Table
1. That is, a positively charged cluster and a non-polar surface
around Tyr 1 in cyt f (i, ii), and two negatively charged clusters
and a non-polar surface surrounding the copper-atom in pc (iii, iv).
Fig. 1. Molecular
detail of the structural sites of oxidation-reduction in the complexes
between cytochrome f and plastocyanin (cyt f-pc).
In
order to facilitate their visualization,
the heme macrocycle in cyt f and the copper-atom in pc are drawn
in 'ball and stick' sizes higher
than in the surrounding amino acid residues. Color code:
light blue, iron-atom in cyt f; red,
copper-atom in pc; yellow, heme macrocylce in cyt f.
| cyt f pc |
(i) Positively charged
cluster constituted of
Lys58,a Lys65, Lys185, Lys187, Arg209 (ii) Non-polar surface around Tyr 1 including Iso3, Phe4, Leu61, Ala62, Pro117, Tyr160, Pro161 (iii) Two negatively charged clusters constituted
of
|
top
3.2. Structural
pathways of electron transfer in cyt f-pc
In higher plants chloroplasts the cytochrome b6f (cyt b6f)
complex transfers electrons between the photosystem II complex and the
P700 reaction center in the photosystem I (PSI) complex. From cytochrome
f (cyt f) in cyt b6f to PSI, the electrons are shuttled by the mobile redox
carrier plastocyanin (pc) [3-6]. Fig. 2 shows a detail
of the electron transfer interface in the cyt f-pc complex obtained from
molecular modelling of the cyt f-pc interaction. In short, electron
tranfer between the Fe-atom in the cyt f heme and the tyrosine 83 (Y83)
in plastocyanin starts with the oxidation of the Fe-atom and electron transfer
directly to the OH group or the pi-electron system of tyrosine 1 (Y1) in
cyt f, or through an intermediate pyrrole ring in the cyt f heme macrocycle.
From there, the electron transfer route goes through the Cu-coordination
centre following most likely one or several molecular pathways.
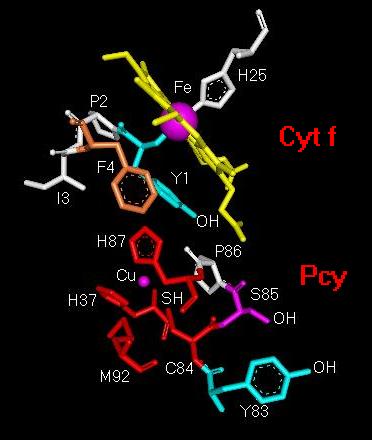
Fig. 2. Structural
detail of the molecular electron-transfer pathway from
the Fe-atom in cytochrome f (Cyt f)
to tyrosine 83 in plastocyanin (Pcy).
Abbreviations: Cu, copper atom in Pcy; C84, cysteine
84; Fe, iron atom in cyt f
heme; F4, phenylalanine 4; H25, histidine 25; H37, histidine
37; H87, histidine
87; I3, isoleucine 3; M92, methionine 92; P2, proline
2; P86, proline 86; S85,
serine 85; Y1, tyrosine 1; Y83, tyrosine 83.
A schematic representation of the distances between the amino acid residues that most likely participate in electron transfer in the cyt f-Pc complex is given in Fig. 3. The figure shows that the distance from the Fe-coordination centre to Y1 is approximately 7.2 Å and 5.9 Å from Y1 to the Cu-ion. From C84(SH) to S85(OH) and from S85(OH) to Y83(OH) the electron transfer distances are respectively about 7.0 Å and 5.2 Å. The figure indicates also that another possible electron transfer route is between C84(SH) and the p-electron system in Y83 with an interaction distance of the order of 7.3 Å. This interesting since it has been emphasized in past discussions that electron transfer distances higher than about 5 Å are a reliable indication of electron transfer occurring by tunnelling efffect, that is, by a quantum jump from one electronic state to another (see, e.g., [1]).
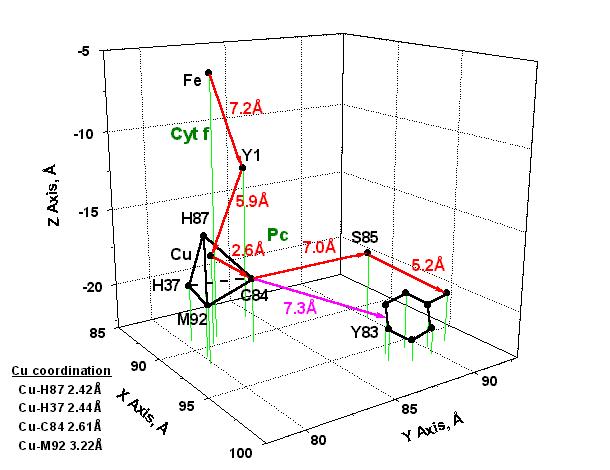
Fig. 3. Model of electron transfer from the
Fe-coordination centre in cytochrome f
(Cyt f) to the Cu-coordination centre and
tyrosine 83 in plastocyanin (Pc).
The
X, Y, Z
coordinates were obtained from the pc structure shown in Fig. 1.
The most probable pathways
of electron transfer are shown with red
arrows;
another possible route of electron transfer
between C84 and the p-electron system in Y83
is indicated with a magenta arrow.
Abbreviations:
Cu, copper ion in Pc; C84, cysteine 84;
Fe, iron ion in cyt f heme; H37,
histidine 37; H87, histidine 87; M92, methionine 92;
S85, serine 85; Y1, tyrosine 1;
Y83, tyrosine 83.
Fig. 3 indicates also
that the distance Cu-C84(SH) is approximately 2.6 Å, i.e., the coordination
distance from the Cu-ion to the SH group in C84. This is a quite
small electron transfer distance in relation to the 5 to 7 Å determined
for the other pathways illustrated in the figure. At first,
this is a convincing argument to suggest the co-existence of different
molecular mechanisms of electron transfer in the Cyt f-Pc complex.
Secondly, an electron transfer distance of about 2.6 Å is a good
indication that in the Cu-coordination centre, and maybe also in its molecular
nearness, the electron transfer reaction originates in van der Waals (vdW)
contacts. This question is discussed further in section (ii) below.
Fig.
3 indicates also that the Cu-ion in pc is coordinated to the
cysteine 84 (C84) thiol group, the methionine 92 (M92) thioether
group, and the histidines 37 (H37) and 87 (H87) imidazole groups in
a quite highly distorted tetrahedral configuration. The corresponding
coordination distances are given in the figure. Such unstable
three-dimensional configuration has partly its origin in differences of
conformational strain energy (DHs) of the amino
acid residues (see discussion in [25]) in the Cu-coordination centre.
That is, DHs = 0.08 (C84), 11.68 (M92)
and 14.15 (H37, H87) kJ/mol. This materializes in the almost identical
coordination distances for Cu-H87 (2.42 Å), Cu-H37 (2.44 Å)
and Cu-C84 (2.61 Å) as compared to the quite different Cu-M92 distance
(3.22 Å) which gives thus rise to the distortion observed in the
coordination tetrahedron.
The distorted tetrahedral configuration of the Cu-coordination
centre is obviously unstable from a thermodynamic viewpoint, inusmuch as
the copper ion may adopt two different configurations; that is, the
configurations corresponding to the coordination numbers (CN) four or six
(see, e.g., [26]). It is worth noting at this point that a
fifth and a sixth coordination partners corresponding to CN = 6 are predicted,
in addition to the more common four coordination partners of the Cu-ion
in pc which are usually identified with the amino acid residues described
above. The implicit fifth and sixth coordination partners were not,
however, correctly identified. Nevertheless, this matter raises an
interesting point. In fact, the crystal radii for the Cu-ion being
0.74 Å for CN = 4 and 0.91 Å for CN = 6 [26], it becomes
perceptible that any molecular volume change in the Cu-centre should
have functional consequences that have not yet been determined, but which
are certainly instrumental in influencing the electron transfer rate in
cyt f-pc, at least in the route from the Cu-ion to Y83. This may
take place, for example, by changing the arrangement and the reorganizational
energy [1] of the van der Waals contacts in the region comprising
the H87
imidazole ring, the Cu-ion and the C84 thiol group. This means
that supplemental electron transfer-induced dynamical distortion of the
already distorted tetrahedron shall thereby create an oscillatory unstable
state upon electron transfer that may render the copper coordination center
a highly unstable region of the plastocyanin molecule which is exactly
what is suitable for rapid electron transfer excursion sequences.
top
3.3.
Intramolecular electron transfer: Tunnelling effect
The theoretical prediction of the
probability of an electron penetrating an energy barrier, that is, the
electron tunnelling through a barrier has been tried in many previous works
(see discussions in [1,2]). Various mathematical expressions
were used to determine the
tunnelling rate constant [1,2,27-37],
kd (in s-1). However, comparison of the theoretical
results with experimental data showed that quite often that the observed
results are usually smaller, sometimes by several orders of magnitude,
than those predicted by theory. For
example, the rate constants of plastocyanin reduction
were shown to be in most cases between about 2800 and 8200 s-1
(see, e.g., [31,38,39]), whereas the values obtained theoretically
are generally much higher. These discrepancies stem partly from the
complex structure of the molecular pathway followed by the electron in
its flight from the electron donor site (EDS) to the acceptor site (EAS).
Additional complications result from the dynamical changes accompanying
the electron transfer either at EDS or at EAS, or in-between, which obviously
change the energy states of the various molecular steps in the structural
pathway of electron transfer. These questions have been approached
in some instances (see discussions on, e.g, the energy of reorganization
in [1,2]), but so far the mathematical expressions of kd
are not yet completely adequate to give a reasonable fit between theory
and experiment. The modification of the mathematical expressions
of kd to include the structural and molecular dynamics parameters
above mentioned is an interesting task that deserves to approached in future
works. This may be achieved using, for example, the new methods
of the knowledge-based models of simulate the redox proteins core at the
molecular sites, or regions, where an electron in a local pool of 'nEDS
electrons' is displaced to a pool of 'mEAS electrons'
by either a non-adiabatic (e.g., a tunnelling jump) or an adiabatic process
of the kind extensively discussed in [1,2,40].
In spite of the afore discussed difficulties, most of the theoretical expressions mentioned in [1,2,27-37] were shown to be instrumental in the study of several aspects of the electron tunnelling in proteins involved in biological oxidation-reduction, and also in electron trasfer reactions via Van der Waals molecular contacts. In this work , the tunnelling rate constant, kd , for a one-dimensional square barrier was determined according to the expression of McElroy et al. [30] (eq. 1).
kd = [16E2(V-E)/hV2] exp{-2p[8m(V-E)]1/2/h]d} eq. 1
where h is Planck's constant, m the mass of the electron, V the barrier height above the electron energy, i.e., potential energy of the electron inside de barrier, E the kinetic energy of the approaching electron (outside the barrier), and d the tunnelling distance. In eq. 1, kd is the probability of an electron crossing the barrier by tunnelling, and in such sense it is the tunnelling rate constant given in s-1. Eq. 1 was transformed to give the values of kd as a function of V, dV (= V-E) and d, for a particular electron transfer pathway. Taking into account that h = 6.626196 x10-34 J.s, m = 9.109558 x 10-31 kg, d is given in Å units, and E, V and dV are given in eV, one has
kd = 3.863 x 1015 [1-(dV/V)]2dV exp[-1.026(dV1/2)d] eq. 2
The evolution of kd as a function of concomitant variations of dV and V is seen in the three-dimensional graphical representations displayed in Fig. 4. To construct the graphs in this figure, the distances d determined in Fig. 2 were used, that is, d = 7.2 Å in the electron transfer route from the iron-atom to the tyrosine 1 (Y1) in cytochrome f, i.e., the Fe(cyt f) ® Y1-OH(cyt f) route (A), and d = 5.9 Å in the electron transfer route from Y1 to the copper-atom in platocyanin, i.e., the Y1-OH(cyt f) ® Cu(pc) route (B).
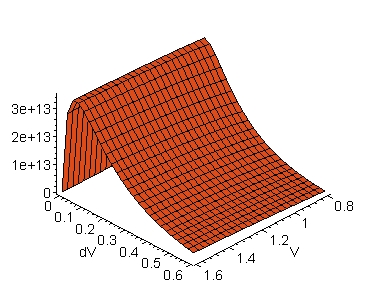
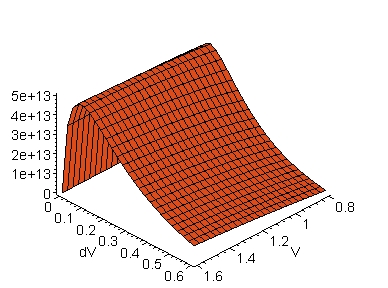
A: Fe(cyt f) ® Y1-OH(cyt f) B: Y1-OH(cyt f) ® Cu(pc)
Fig. 4. Tunnelling rate constant, kd (in
s-1), along the structural pathways of electron transfer in
the cytochrome f-
plastocyanin complex. The Y axes are the tunnelling rate
constants calculated according to eq. 2 (see text). The distances,
d (in Å),
between the electron donor-acceptor sites were obtained from the molecular
coordinates of the cyt f-pc complex as is shown in Fig. 3;
d is 7.2 Å in the Fe(cyt f) ®
Y1-OH(cyt
f) route (A), and 5.9 Å
in the Y1-OH(cyt f) ® Cu(pc)
route
(B). Abbreviations:
cyt f,
cytochrome f; Cu(pc), copper-atom in pc; Fe(cyt f), iron-atom
in cyt f; pc, plastocyanin; Y1, tyrosine 1.
An interesting observation in Fig. 4 is that the
kd maxima are observed for dV's of the order of 0.1 eV for any
V in the range from 0.8 to 1.6 eV. This is aggreement with the notion
that the energy differences between V and E (= dV) might, in general, be
small (see, e.g., [2]). It is important to note, however,
that the dV value at kd maximum, dVkd(max), decreases
with d (Fig. 5) according to an exponential decay as is seen in
Fig.
6. This is also illustrated below in eq. 3.
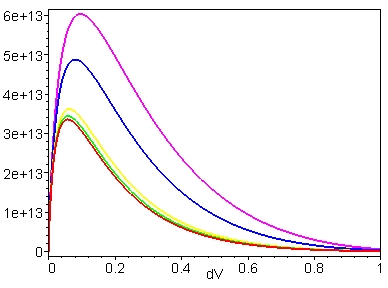
Fig. 5. Tunnelling rate constant, kd (in
s-1), as a function of dV for the structural pathways
of electron transfer in the cytochrome f-plastocyanin complex. The
Y
axis is the tunnelling rate
constant calculated according to eq. 2 (see text). The distances,
d (in Å), between the electron donor-
acceptor sites were obtained from the molecular coordinates of the cyt
f-pc complex as is shown in
Fig. 3. Color code for the distances d: magenta,
5.2 Å; blue, 5.9 Å; yellow,
7.0 Å; green, 7.2 Å;
red, 7.3 Å. Abbreviations: cyt f, cytochrome
f; pc, plastocyanin.
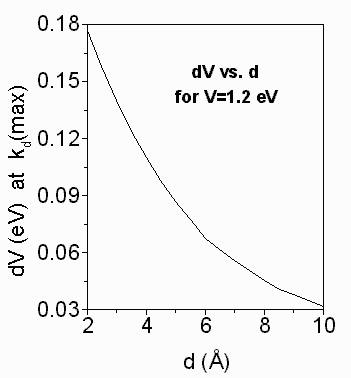
Fig. 6. dV (in eV) at the kd maximum
(in s-1) as a function of d (in Å). dV
is given by the expression
dV=0.011 + 0.167exp[-(d-2)/3.786]. The dV's (dV=V-E) and the distances,
d (in Å), between the electron
donor-acceptor sites and were were calculated according eq.
2 (see text). Abbreviations: cyt f, cytochrome
f;
d, tunnelling distance; dV=V-E; E, kinetic energy of the electron
approaching the barrier; kd, tunnelling rate
constant; pc, plastocyanin; V, barrier height above the electron
energy.
The exponential decay of dVkd(max) with increasing d (Fig. 6) is represented by the expression
dVkd(max) = 0.011 + 0.283exp(-0.264d), eq. 3
The limiting values in eq. 3 are
0.294 eV > dVkd(max) > 0.178 eV, for
d < 2 Å,
0.178 eV > dVkd(max) > 0.011 eV, for
2 Å < d < ~20-30 Å, and
dVkd(max) ~ 0.011 eV, for
d > ~20-30 Å.
An interesting point in this respct is that at short distances
between the electron donor and acceptor sites, the dVkd(max) difference
may take much larger values than the dVkd(max) differences for
donor and acceptor sites separated by large distances, where dV has to
be small for an efficient electron transfer to take place. In other
words, an efficient electron transfer at the most usual tunnelling distances,
i.e., from 5 to 10 Å (see, e.g., [1]), implies that the energy
of the electron approaching the tunnelling barrrier, E, must be close to
the energy of the tunnelling barrier, V, or almost identical (see discussions
in [2]).
top
3.4. Intramolecular electron
transfer: Van der Waals contact
From the above considerations, it is reasonable to foresee
that the transfer of an electron from Y1 (in cyt f) to the Cu-ion in plastocyanin,
the bond lengths in the Cu-coordination centre must distort from their
steady-state geometry, or quasi-equilibrium condition, to a distorted
geometry, or transition-state geometry, where lenghtening and contraction
of bonds might occur. For this to take place, energy is expended
prior to the return of the coordinnation centre to its equilibrium geometry.
A structural model to take these effects into account is presented in Fig.
7.
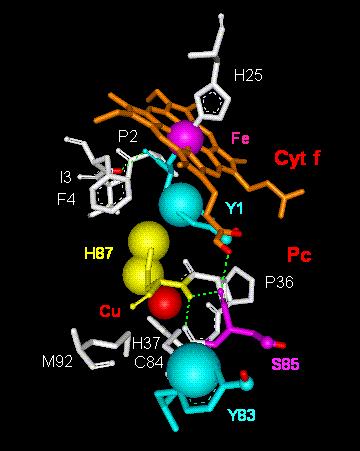
Fig. 7. Model of electron transfer via van der
Waals contacts throughout
tyrosine 1 in cytochrome f (Cyt f) to histidine
87 and the Cu-atom in plastocyanin
(Pc). Abbreviations:
Cu,
copper atom in Pc; C84, cysteine 84; Fe, iron atom in cyt f
heme; F4, phenylalanine 4; H25, histidine 25; H37, histidine
37; H87, histidine 87;
I3, isoleucine 3; M92, methionine 92; P2, proline 2;
P36, proline 36; S85, serine 85;
Y1, tyrosine 1; Y83, tyrosine 83.
Fig. 7 shows the most likely 'van der Waals routes' (vdW1,vdW2) together with the 'tunnelling pathways' (T1-T3) discussed above (Figs. 2 and 3). A summary is given in Scheme I with indication of the respective tunnelling or van der Waals distances in square brackets (cf. also Fig. 3):
| vdW1: Y1(cyt f)®H87(pc)
[... Å]
H87(pc)®Cu(pc) [... Å] vdW2: Cu(pc)®C84(pc) [2.6 Å] |
| T1: Fe(cyt
f)®Y1-OH(cyt f) [7.2 Å]
Y1-OH(cyt f)®Cu(pc) [5.9 Å] T2: C84(pc)]®S85(pc) [7.0 Å] S85(pc)®Y83-OH(pc) [5.2 Å] T3: C84(pc)]®Y83[p-electron system](pc) [7.3 Å] |
Now, the various structural rearrangements and molecular dynamics in these intramolecular pathways during electron transfer from the iron-atom in cytochrome f to the copper-atom in plastocyanin are certainly at least some of the factors that may have to taken into account for the correction of mathematical expressions of kd as, for example, McElroy et al.'s equation [30] used in this work (eq. 1). A reasonable correction, although empirical at this stage, would be to multiply kd by an appropriate scale factor, f, that is,
kd = [16E2f(V-E)/hV2] exp{-2p[8m(V-E)]1/2/h]d} eq. 4
For example, an appropriate accord between the theoretical and experimental kd's for the reduction of plastocyanin by cytochrome f would be obtained for f values from 0.5 to 20 x 10-10, meaning that eq. 2 would take the lower and upper limits represented by eqs. 5 and 6, that is,
kd = 1.931 x
105 [1-(dV/V)]2dV exp[-1.026(dV1/2)d]
eq. 5
and kd = 77.260 x
105 [1-(dV/V)]2dV exp[-1.026(dV1/2)d]
eq. 6
This would yield kd(max) between 2200 and 84000 s-1
(cf., in this respect, [31,38,39]) for barrier heights, V, and tunnelling
distances, d, of the order of 0.8 eV and 6 Å, respectively.
It is acknowledge, however, that it is not yet possible to correlate this
still empirical correction to the molecular dynamics (e.g., structural
rearrangements and/or side chains movements) of electron transfer occurring
at the docking interfaces or inside the protein core of two biological
redox macromolecules, here the cytochrome f and plastocyanin molecules.
top
4. Concluding remarks
First, the data shown in Figs. 2, 3 and 7 indicate that different electron transfer mechanisms may take place in the same cyt f-pc complex, that is, electron transfer by tunnelling effect and via van der Waals molecular contacts. This may have far reaching consequences since, in a first approximation, it presupposes the parallel function of adiabatic and non-adiabatic electron transfer processes, thereby implying the interplay of mechanisms with different temperature dependence, or requirements [1,2]. The coexistence of the tunnelling effect and the Van der Waals contacts in the same electron transfer rapid pennetrative excursion may well constitute a means of control of the physiological function of the cyt-Pc complex, but also of the electron transfer reactions between the cytochrome b6f complex and P700, i.e., the reaction center of photosystem I in the photosynthetic membrane.
Secondly, another question still remaining is the possibility that water molecules, or the displacement of hydrogen ions, may provide an efficient mechanism and pathway of electron transfer (see discussions in [40,41]). In which the cyt f-Pc complex is concerned, the mediation or facilitation of electron transfer by interposed water molecules inside the protein complex is an assumption that, though attractive, has most probably to be ruled out on account of arguments discussed in computer simulations of water-plastocyanin interaction energies [42]. Nevertheless, a recent work of Sainz et al. [43] shows that (i) an internal chain of five H2O molecules in the protein core of cyt f near the heme macrocycle just before the docking interface is important to its function, and (ii) the perturbation of this chain of water molecules impairs the cytochrome f function. In addition, there is evidence indicating that proton transfer to a buried redox center in proteins is a type of mechanism that may be instrumental in electron transfer [44].
Finally, it is implicit in the pathways schematized
in Figs. 2 and 7 that one needs to
modify the mathematical expressions of kd (eqs. 1-4)
in order to include structural and molecular dynamics parameters such as,
e.g., the effect of
side chain movements on electron transfer through
'electron transfer holes' such as those in the cyt f-pc complex.
This is an interesting task that shall be undertaken
using, for example, knowledge-based models (see recent work
in [45]) to simulate the molecular core of the redox proteins at
the sites, or molecular domains, where the electron displacements take
place by either non-adiabatic tunnelling jumps or van der Waals processes
[1,2,46].
top
5. Abbreviations
| cyt b6f
cyt f d dV dVkd(max) E kd pc P700 PSI PSII V vdW |
cytochrome b6f
cytochrome f distance between the electron donor and acceptor sites (in Å) = V - E (in eV) dV at the kd maximum (in eV) [cf. eqs. 4 and 5] kinetic energy of the electron approaching the barrier (in eV) tunnelling rate constant (in s-1) plastocyanin reaction center in PSI photosystem I photsystem II barrier height above the electron energy (in eV) van der Waals |
top
Acknowledgements
This work was supported by grant OGP0006357 from the Natural
Science and Engineering Research Council of Canada and institutional grants
from the Université du Québec à Trois-Rivières,
and is the result of a collaboration with Dr. A. Kajava at the Institut
Suisse de Recherches Expérimentales sur le Cancer (ISREC), Groupe
de Bioinformatique, Epalinges s/Lausanne, Suisse. I wish to thank
Dr. A. Kajava and the Bioinformatics Group and Library staff at ISREC for
the friendliness of their welcome and help. Last but not least, I
am very grateful to Dr. I. Gabashvili for the many clever comments and
suggestions made during the preparation of this work.
[ 1] D. DeVault, Quantum-mechanical Tunnelling in Biological
Systems, 2nd ed., Cambridge University Press, Cambridge, 1984.
[ 2] R.A. Marcus, N. Sutin, Electron transfer in chemistry
and biology, Biochim. Biophys. Acta 811 (1985) 265-322.
[ 3] H. Bohner, H. Böhme, P. Böger, Reciprocal
formation of plastocyanin and cytochrome c-553 and the influence of cupric
ions on
photosynthetic electron
transport, Biochim. Biophys. Acta. 592 (1980) 103-112.
[ 4] G. Sandmann, Reck., E. Kessler, P. Böger, Distribution
of plastocyanin and soluble plastidic cytochrome c in various classes of
algae,
Arch. Microbiol. 134
(1983) 23-27.
[ 5] K.K. Ho, D.W. Krogmann, Electron donors to P700 in
cyanobacteria and algae. An instance of unusual genetic variability,
Biochim.
Biophys. Acta. 766
(1984) 310-316.
[ 6] H. Zhang, H.B. Pakrasi, J. Whitmarsh, Photoautotrophic
growth of the cyanobacterium Synechocystis sp. PCC6803 in the absence of
cytochrome c553 and
plastocyanin, J. Biol. Chem. 269 (1994) 5036-5042.
[ 7] G. Sandmann, P. Böger, Physiological factors
determining the formation of plastocyanin and plastidic cytochrome c-553
in
Scenedesmus, Planta
147 (1980) 330-334.
[ 8] S. Merchant, L. Bogorad, Metal ion regulated gene
expression: use of a plastocyanin-less mutant of Chlamydomonas reinhardtii
to
study the Cu(II)-dependent
expression of cytochrome c-552, EMBO J. 6 (1987) 2531-2535.
[ 9] C. Frazão, C.M. Soares, M.A. Carrondo, E. Pohl,
Z. Dauter, K.S. Wilson, M. Hervás, J.A. Navarro, M.A. De la Rosa,
G.M. Sheldrick, Ab
initio determination
of the crystal structure of cytochrome c6 and comparison with plastocyanin,
Structure 3 (1995) 1159-1169.
[10] E.L. Gross, Plastocyanin: structure, location, diffusion,
and electron transfer mechanisms. in: D. Ort, C. Yokum (Eds.), Oxygenic
Photosynthesis:
The Light Reactions, Kluwer Academic Publishers, Dordrecht, The Netherlands,
1996, pp. - .
[11] T. Takabe, H. Ishikawa, S. Niwa, Y. Tanaka, Electron
transfer reactions of chemically-modified plastocyanins with P700 and
cytochrome f,
J. Biochem. (Tokyo) 96 (1984)385-393.
[12] G.P. Anderson, D. G. Sanderson, C. H. Lee, S. Durell, L.
B. Anderson, E. L. Gross, The effect of ethylenediamine chemical
modification of plastocyanin
on the rate of cytochrome f oxidation and P700 reduction, Biochim. Biophys.
Acta 894 (1987) 386-396.
[13] L.Z. Morand, M.K. Frame, K.K. Colvert, D.A. Johnson, D.W.
Krogman, D.J. Davis, Plastocyanin-cytochrome f interaction,
Biochemistry 28 (1989)
8039-8047.
[14] T. Takabe, H. Ishikawa, Kinetic studies on a cross-linked
complex between plastocyanin and cytochrome f, J. Biochem. (Tokyo) 105
(1989) 92-102.
[15] S. Modi, S. He, J.C. Gray, D.S. Bendall, The role of surface
exposed tyr-83 of plastocyanin in electron transfer from cytochrome c,
Biochim. Biophys.
Acta. 1101(1992) 64-68.
[16] S. Modi, M. Nordling, L.G. Lundberg, Ö. Hansson, D.S.
Bendall, Reactivity of cytochromes c and f with mutant forms of spinach
plastocyanin, Biochim.
Biophys. Acta 1102 (1992) 85-90.
[17] L. Qin, N.M. Kostic, Importance of protein rearrangement
in the electron-transfer reaction between the physiological partners
cytochrome f and plastocyanin,
Biochemistry 32 (1993) 6073-6080.
[18] T.B. Karpishin, M.W. Grinstaff, S. Komar-Panicucci, G. McLendon,
H.B. Gray, Electron transfer in cytochrome c depends upon the
structure of the intervening
medium, Structure 2 (1994) 415-422.
[19] F.C. Bernstein, T.F. Koetzle, G.J.B. Williams, E.F. Meyer,
M.D. Brice, J.R. Rogers, O. Kennard, T. Shimanouchi, M. Tatsumi, The
Protein Data Bank:
a computer-based archival file for macromolecular structures, J. Mol. Biol.
112 (1977) 535-542.
[20] H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat,
H. Weissig, I.N. Shindyalov, P.E. Bourne. The Protein Data Bank,
Nucleic Acids Res.
28 (2000) 235-242.
[21] A. Roussel, C. Cambillan, In Silicon Graphics Geometry Partner
Directory (Fall 1989), Silicon Graphics, editor, Silicon Graphics,
Mountain View, CA.
pp 77-78.
[22] A. Roussel, A.G. Inisan, TURBO-FRODO, Version 4.3, release
a. Bio-Graphics, Marseille, France, 1993.
[23] A.T. Brunger, X-PLOR Version 3.1: A System for X-Ray Crystallography
and NMR. Yale University Press, New Haven, CT, 1992.
[24] A.V. Kajava, Modeling of a five-stranded coiled coil structure
for the assemby domain of the cartilage oligomeric matrix protein,
Proteins: Structure,
Function and Genetics 24 (1996) 218-226.
[25] K. Sak, M. Karelson, J. Jarv, Bioorg. Chem. 27 (1999) 434-442.
[26] B. Douglas, D.H. McDaniel, J.J. Alexander, Concepts and
Models of Inorganic Chemistry, 2nd ed., John Wiley, New York, 1983.
[27] D. DeVault, B. Chance, Biophys. J.
6 (1966) 825-.
[28] T. Kihara, B. Chance, Cytochrome photooxidation
at liquid nitrogen temperatures in photosyhthetic bacteria, Biochim. Biophys.
Acta
189 (1969) 116-124.
[29] J.J. Hopfield, Electron transfer between
biological molecules by thermally activated tunneling, Proc. Nat. Acad.
Sci. USA 71 (1974)
3640-3644.
[30] J.D. McElroy, D. Mauzerall, G. Feher,
Characterization of primary reactants in bacterial photosynthesis.
II. Kinetic studies of the
light-induced EPR signal (g = 2.0026) and the optical absorbance changes
at cryogenic temperatures, Biochim. Biophys. Acta 333
(1974) 261-177.
[31] M. Fragata, F. Bellemare, Dielectric
constant dependence of biological oxidation-reduction. 1. A model
of polarity-dependent
ferrocytochrome c oxidation, Biophys. Chem. 15 (1982) 111-119.
[32] G. McLendon, R. Hake, Interprotein
electron transfer, Chem. Rev. 92 (1992) 481-490.
[33] J.M. Nocek, J.S. Zhou, S. De Forest,
S. Priyadarshy, D.N. Beratan, J.N. Onuchic, B.M. Hoffman, Theory and practice
of electron
transfer within protein-protein complexes: application to the multidomain
binding of cytochrome c by cytochrome c peroxidase,
Chem. Rev. 96 (1996) 2459-2489.
[34] A.J.A. Aquino, P. Beroza, J. Reagan,
J.N. Onuchic, Estimating the effect of protein dynamics on electron transfer
to the special pair in
the photosynthetic reaction center, Chem. Phys. Let. 275 (1997) 181-187.
[35] M.R. Haris, D.J. Davis, B. Durham,
F. Millett, Temperature and viscosity dependence of the electron-transfer
reaction between
plastocyanin and cytochrome c labeled with a ruthenium(II) bipyridine complex,
Biochim. Biophys. Acta 1319 (1997) 147-154.
[36] C.A. Cunha, M.J. Romão, S.J.
Sadeghi, F. Valetti, G. Gilardi, C.M. Soares, J. Biol. Inorg. Chem. 4 (1999)
360-374.
[37] G.N. Chuev, V.D. Lakhno, M.N. Ustitnin,
Superexchange coupling and electron transfer in globular proteins via polaron
excitations,
J. Biol. Phys. 26 (2000) 173-184.
[38] L. Qin, N.M. Kostic, Importance of
protein rearrangement in the electron-transfer reaction between the physiological
partners
cytochrome f and plastocyanin, Biochemistry 32 (1993) 6073-6080.
[39] Y. Ilan, A. Shafferman, Biochim. Biophys. Acta 501 (1978
127.
[40] S.T. Prigge, A.S. Kolhekar, B.A. Eipper, R.E. Mains, L.M.
Amzel, Nature Struct. Biol. 6 (1999) 976-983.
[41] G.N.R. Tripathi, J. Am. Chem. Soc. 120 (1998) 4161-4166.
[42] C.X. Wang, S. Cannistraro, Il Nuovo Cimento 5 (1985) 405-414.
[43] G. Sainz, C.J. Carrell, M.V. Ponamarev, G.M. Soriano, W.A.
Cramer, J.L. Smith, Interruption of the internal water chain of
cytochrome f impairs
photosynthetic function, Biochemistry 39 (2000) 9164-9173.
[44] K. Chen, J. Hirst, R. Camba, C.A. Bonagura, C.D. Stout,
B.K. Burgess, F.A. Armstrong, Atomically defined mechanism for proton
transfer to a buried
redox centre in a protein, Nature 405 (2000) 814-817.
[45] J. Xiong, S. Subramaniam, Govindjee, Aknowledge-based three
dimensional model of the Photosystem II reaction center of
Chlamidomonas reinhardtii,
Photosynth. Res. 56 (1998) 229-254.
[46] M.V. Volkenstein, General Biophysics,
Vol. II, Academic Press, New York, 1983.
| Mário Fragata
fragata@uqtr.uquebec.caNovember 14, 1967 to gsi at 8:15am |