Ground and Singlet Excited State Hydrogen-Bonding
Interactions between 1-Azacarbazole and Amides
Manuel Galán, Carmen Carmona, Pilar Guardado, María A. Muñoz
and Manuel Balón.*
Departamento de Química Física, Facultad de Farmacia,
Universidad de Sevilla, 41012 Sevilla, Spain.
Abstract
In cyclohexane as the solvent, 1-azacarbazole (or a -carboline), AC, forms fluorescent ground state 1:1 hydrogen-bonded
complexes with N,N-dimethylformamide, DMF, N,N-dimethylacetamide, DMA, and hexamethylphosphoramide,
HMPA. The absorption and fluorescence spectra of the complexes are red shifted
with respect to those of the no-bonded AC, and their association constants increase
as the hydrogen-bonding acceptor properties of the amides increase. Fluorescence
of the AC-DMF and AC-DMA solutions show mono- or biexponential decays depending
on the monitored emission wavelength. To aid the interpretation of these results
we have also studied the effect that triethylamine and methylethylcetone addition
produces on the absorption and fluorescence spectra of AC. Triethylamine does
not significantly affect the absorption spectrum of AC, but it dynamically quenchs
its fluorescence. Methylethylcetone, conversely, behaves as amides do. On the
basis of the above results, we assume that, in the ground state, the hydrogen
bonding interaction takes place between the pyrrolic NH group of AC and the
carbonyl group of the amide. Hydrogen bonded complexes and no-bonded AC behave
in the singlet excited state as independent fluorophores. Singlet excited state
of free AC is dynamically quenched by DMF and DMA. The quenching mechanism involves
the hydrogen-bonding interaction of the pyrrolic NH group of AC and the lone
electron pair of the amide. For HMPA, probably due to geometrical restrictions,
this quenching process is absent.
Keywords: 1-azacarbazole, amides, hydrogen-bonding, ground state, excited
state.
* Author for correspondence (E-mail: balon@fafar.us.es)
1. Introduction
1-Azacarbazole, or a -carboline, is a representative
member of the carboline family, a class of drug-binding alkaloids possessing
interesting photophysical and biological properties [1]. Because of its structural
relation with 7-azaindole, AI, a molecule whose dimer has long been recognized
as a model for the DNA base pairs, the photophysics of AC has received much
attention. Similarly to AI, AC forms doubly hydrogen-bonded dimers that, upon
photoexcitation, undergo a concerted intermolecular excited state double proton
transfer reaction, ESDPT. Excited-state tautomerization reactions of AI and
AC dimers have been intensively studied as model systems for understanding the
photoinduced mutations of the DNA base pairs [2].
Owing to its bifunctional proton donor and acceptor properties, AC may form
a variety of complexes with different hydrogen-bonding partners. 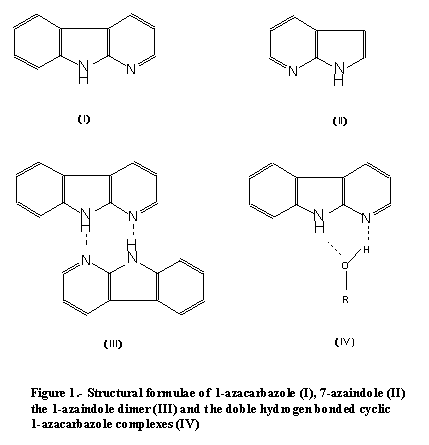 Particularly, cyclic doubly hydrogen-bonded
complexes of the sort illustrated in Fig. 1, have arisen great interest, since
as the AC dimers, they are also able to undergo ESDPT reactions. While much
work has been done on the photophysics of the cyclic complexes [3-12], single
non-cyclic 1:1 hydrogen-bonded complexes involving only one of the hydrogen-bonding
centres of the AC ring have received very scarce attention. However, the study
of the nature of the last complexes and the dynamics of their hydrogen-bonded
mediated proton transfer reactions is key for the full understanding of the
photophysics and photochemistry of AC. Thus, it would be of interest to the
photophysics of these hydrogen-bonded complexes of AC.
Particularly, cyclic doubly hydrogen-bonded
complexes of the sort illustrated in Fig. 1, have arisen great interest, since
as the AC dimers, they are also able to undergo ESDPT reactions. While much
work has been done on the photophysics of the cyclic complexes [3-12], single
non-cyclic 1:1 hydrogen-bonded complexes involving only one of the hydrogen-bonding
centres of the AC ring have received very scarce attention. However, the study
of the nature of the last complexes and the dynamics of their hydrogen-bonded
mediated proton transfer reactions is key for the full understanding of the
photophysics and photochemistry of AC. Thus, it would be of interest to the
photophysics of these hydrogen-bonded complexes of AC.
In this context, we have carried out a spectroscopic study (uv-vis, steady-state
and time-resolved fluorescence) of the ground and singlet excited state hydrogen-bonding
interactions of AC with different amides; namely, N,N-dimethylformamide, DMF,
N,N-dimethylacetamide, DMA, and hexamethylphosphoramide, HMPA. These amides
possess only proton accepting groups, hence they can only form hydrogen-bonds
with the pyrrolic NH site of the AC. These studies have been conducted in cyclohexane,
a solvent that due to its low polarity and its negligible hydrogen-bonding properties
can be considered inert.
2. Experimental
1-Azacarbazole was synthesized and purified as described in the literature
[13]. The hydrogen-bonding acceptors DMF, DMA, and HMPA, triethylamine, methylethylcetone
and the cyclohexane used as the solvent were commercial products (Sigma, Aldrich)
of the best available quality, they were used without further purification and
were stored on 4 Å molecular sieves.
The uv-visible absorption spectra were recorded on a Perkin-Elmer Lambda-5
spectrophotometer. Stationary fluorescence measurements were carried out in
a Hitachi F-2500 spectrofluorimeter. Fluorescence lifetimes were measured with
an Edinburgh Analytical Instruments CD-900 spectrofluorimeter employing the
time correlated single photon counting technique. The source was a nanosecond
flashlamp filled with H2 (0.4 bar) opperating at 6.8 kV, with a repetition
rate of 40.0 kHz. Fluorescence decays were acquired to 104 counts
in the peak. The decays were repeated at least twice and were fitted by reference
deconvolution to a sum of exponentials:
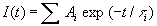
with amplitudes, Ai, and lifetimes, t i. Decay curves were both individually and globally
analyzed by using single, double and triple exponentials. Goodness of the individual
fits was judged by the magnitude of c 2r
and the shape of the autocorrelation function of the weighted residuals.
The analysis of the lifetime data at different acceptor concentrations were
performed with a global analysis program based on the Marquardt algorithm. These
results were judged by the statistical fitting parameter c 2g. Fluorescence measurements were
carried out with nondegassed solutions under temperature controlled conditions
(25±0.1 ºC).
3. Results and Discussion
As stated in the introductory section, AC is prone to dimerize in low polar
solvents. To check this possibility, we previously studied the concentration
dependent absorption and fluorescence spectra of AC in cyclohexane. In the AC
concentration range used in this work, up to 10-4 M, we did not observe
changes in the absorption spectrum attributable to dimer formation and the absorbances
fitted acceptably the Lambert-Beer law at different absorption wavelengths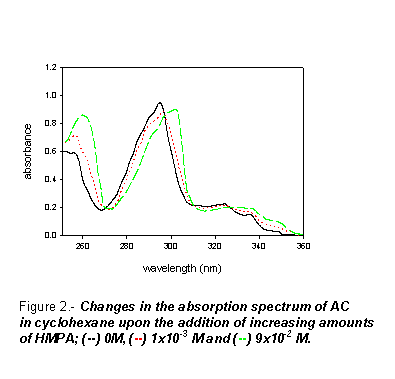 . Neither the fluorescence spectrum of AC
showed indication of dimer formation. The dimer would be clearly detected by
the emission of its photoinduced tautomer at 500 nm, which was absent in the
fluorescence spectra of AC in cyclohexane under our experimental conditions.
. Neither the fluorescence spectrum of AC
showed indication of dimer formation. The dimer would be clearly detected by
the emission of its photoinduced tautomer at 500 nm, which was absent in the
fluorescence spectra of AC in cyclohexane under our experimental conditions.
Figure 2 shows the changes produced in the uv-visible and fluorescence spectra
of AC in cyclohexane upon adding HMPA. Similar changes are observed for DMA
and DMF. As this Figure shows, the uv-vis absorption spectrum of AC is shifted
to the red as the amide concentration is increased. The presence of an isosbestic
point in these spectra indicated the formation of AC-amide stoichiometric complexes.
Owing to the nature of the interacting substrates we assume that these complexes
are stabilized by hydrogen bonding interactions involving the pyrrolic NH group
of the AC ring. Unfortunately, the small magnitude of the absorbance changes
precluded the determination of the association constants of the complexes from
the absorption spectra.As it is typically shown in Fig. 3 for the AC-HMPA system,
the fluorescence spectra of the hydrogen bonded complexes of AC with DMA,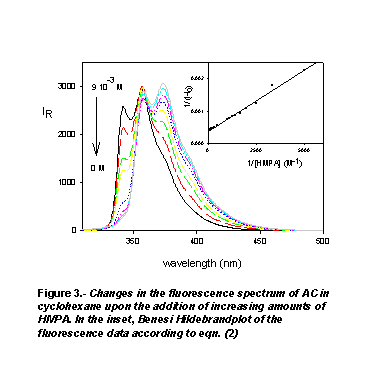 DMF and HMPA are red shifted by 10-15 nm with respect
to that of free AC. The fluorescence data were analyzed using the Benesi-Hildebrand
equation for 1:1 stoichiometric binding,
DMF and HMPA are red shifted by 10-15 nm with respect
to that of free AC. The fluorescence data were analyzed using the Benesi-Hildebrand
equation for 1:1 stoichiometric binding,
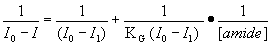
where I0 is the initial fluorescence intensity of free AC at the
titration wavelength, I1 the fluorescence intensity of the AC-amide
complex and I the observed fluorescence intensity of the corresponding AC-amide
mixtures. As the inset in Figure 3 shows, the linear dependence of 1/(I0-I)
on the reciprocal of amide concentration, confirms the assumed 1:1 stoichiometry
of the complexes. The slope and the intercept of these plots allowed us to calculate
the values of the ground state formation constants reported in Table 1. The
sequence of these association constants DMF < DMA < HMPA closely follows
that of b hydrogen-bonding acceptor parameters of
the amides [14]. On the other hand, the red shifts observed in the absorption
and fluorescence spectra upon the formation of the complexes indicate that the
hydrogen-bonding interaction is reinforced in the excited state. This is in
agreement with the charge density decrease experienced by the pyrrole nitrogen
atom upon excitation of AC to its S1 state [12].
Table 1. Ground state association constants, KG,
and dynamic quenching constants, kq,
for the hydrogen-bonding interactions of AC with amides in cyclohexane
at 298 K.
| |
b (a)
|
KG
|
kq
|
|
DMF
|
0.69
|
150
|
13±1
|
|
DMA
|
0.76
|
250±15
|
15±1
|
|
HMPA
|
1.05
|
970±30
|
--
|
(a) Hydrogen-bonding acceptor descriptor values according
Y. Marcus [14].
In order to gain some insight into the dynamics of the hydrogen-bonding interaction
in the singlet excited state, we carried out time-resolved fluorescence measurements
of the AC-amide solutions in cyclohexane. We will discuss firstly the results
obtained for the AC-DMF and AC-DMA systems, since they are somewhat different
from those of the AC-HMPA system. The fluorescence decays of the AC-DMF and
AC-DMA mixtures depended on the emission wavelength. As it is illustrated in
Table 2 for the AC-DMA mixture, at 340 nm the fluorescence decays could be fairly
well described by a linear combination of two exponentials:
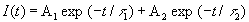
where A1 and A2 stand for the contribution at zero time
of the two components with lifetimes t 1 and t 2.
Table 2. Analysis of the biexponential decays of the
AC-DMA system in cyclohexane at 298 K
( l exc=320 nm, l
em =340 nm). The figures into parenthesis are the pre-exponential
factors, Ai.
|
103 [DMA ]/ M
|
t 1/ns
|
t 2/ns
|
|
2
|
5.6
|
-
|
|
4
|
4.3 (0.062)
|
7.4 (0.021)
|
|
6
|
4.1 (0.063)
|
7.9 (0.016)
|
|
8
|
3.7 (0.056)
|
7.7 (0.018)
|
|
10
|
3.5 (0.052)
|
7.7 (0.018)
|
|
30
|
1.8 (0.046)
|
7.3 (0.033)
|
|
50
|
1.1 (0.036)
|
7.2 (0.036)
|
The shorter lifetimes, t 1,
decreased with the amide concentration, while the longer lifetime component,
t 2, was independent of the amide concentration
and remained practically constant around 7.4 ns.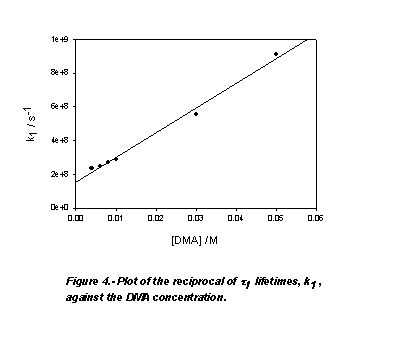 As Fig. 4 shows, the plots of
the reciprocal of the lifetimes, k1=1/t 1, against amide concentration according to
equation
As Fig. 4 shows, the plots of
the reciprocal of the lifetimes, k1=1/t 1, against amide concentration according to
equation
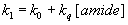
are linears with intercept values, k0, very close to that of the
reciprocal of the lifetime of no-bonded AC in cyclohexane, t
0=6.4 ns. The slopes of these plots, kq, are reported
in Table 1. At the red end of the emission spectra, this short lifetime component
dissapears and the fluorescence decays could reasonably be fitted to monoexponential
functions with lifetimes whose values, within the experimental error, are equal
to those of t 2.
The above results show that, in the excited state, the equilibrium
between the hydrogen bonded complexes and the free AC is not established during
the life span of these species. If this were the case, these species would be
coupled in the excited state and, therefore, the fluorescence decays would be
always biexponential, irrespective of the monitored emission wavelength. On
the other hand, in the case of irreversible formation of the hydrogen-bonded
complexes in the excited state, we would expect biexponential decays at the
longest wavelengths and the appearance of a rising time, i.e. a negative preexponential
factor. Since such behavior was not observed, it is quite unlikely that free
AC is the excited state precursor of the hydrogen-bonded complexes. Therefore
we conclude that, as depicted in Scheme 1, the hydrogen-bonded complexes and
the no-bonded AC behave as independent fluorophores. To account for the dynamic
quenching of the no-bonded AC species by DMF and DMA, we will assume that, the
interaction in the excited state is different than in the ground state.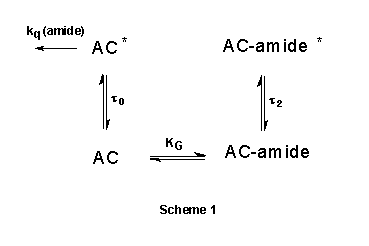
In this sense, it is interesting to realize that DMF and DMA molecules
have two potential acceptor centres for hydrogen-bonding interactions, the oxygen
atom of the carbonyl group and the lone electron pair on the nitrogen atom.
Therefore, it is conceivable that AC could interact independently with each
one of these centres in the amide molecule.
Thus, to obtain information on this point, we have studied the interactions
of AC with triethylamine and methylethylcetone as model compounds. Addition
of triethylamine had not significant effects on the absorption spectrum of AC,
but, as Figure 5 shows, it quenchs the fluorescence intensity and decreases
the fluorescence lifetime of AC.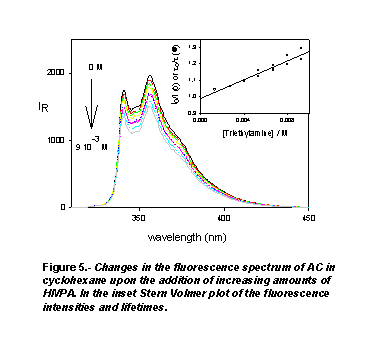 These results show the dynamical nature of
the quenching process; i.e. the quenching is due to the excited state interaction
of AC with the amine. The mean value of the quenching constant obtained from
Stern-Volmer plots of the steady-state and time-resolved fluorescence data,
5x1010 M-1 s-1, is similar to those of the
quenching constants, kq, reported in Table 1 for the AC-DMF and AC-DMA
systems. These quenching constants are, on the other hand, of the order of magnitude
expected for a difussion controlled process in cyclohexane. Conversely, the
changes observed in the absorption and fluorescence spectra of AC in cyclohexane
upon the addition of methylethylcetone entirely resemble those produced by the
amides.
These results show the dynamical nature of
the quenching process; i.e. the quenching is due to the excited state interaction
of AC with the amine. The mean value of the quenching constant obtained from
Stern-Volmer plots of the steady-state and time-resolved fluorescence data,
5x1010 M-1 s-1, is similar to those of the
quenching constants, kq, reported in Table 1 for the AC-DMF and AC-DMA
systems. These quenching constants are, on the other hand, of the order of magnitude
expected for a difussion controlled process in cyclohexane. Conversely, the
changes observed in the absorption and fluorescence spectra of AC in cyclohexane
upon the addition of methylethylcetone entirely resemble those produced by the
amides.
On the basis of the above results, we conclude that, in the ground state, the
pyrrolic NH group of AC interacts preferently with the carbonyl groups of the
amides, but, in the excited state, the hydrogen-bonding interactions take place
through the lone electron pair of the nitrogen atoms of the amides. This excited
state hydrogen bonding interaction efficiently quenchs the AC fluorescence.
Possibly, this interaction involves the transfer of the pyrrolic proton to the
amide and the formation of AC anions which are very weakly fluorescent.
Turning now to the AC-HMPA system, time-resolved fluorescence measurements
showed that, independently of the emission wavelength, the fluorescence of the
cyclohexane solutions of AC in the presence of HMPA decayed monoexponentially.
The lifetimes of the decays were very close to that of no bonded AC, 6.4 ns,
and slightly increased as the acceptor concentration increased. Double exponential
analyses were also attempted, but neither c r2 nor c g2 improved. Furthermore, the second
component of these analyses revealed at random and, occasionally, physically
inexplicable negative contributions to the overall decay. We think that this
conflicting result is an artifact of the fitting procedure which cannot satisfactorily
resolve lifetimes whose values differ less than 1 ns [15]. Thus, the fluorescence
decays of AC-HMPA solutions in cyclohexane could also be reasonably interpreted
according to Scheme 1, assuming a system formed by a mixture of two independently
absorbing and emitting species, the complex and the no-bonded AC, with very
close fluorescence lifetime. Interestingly, HMPA, possibly due to geometrical
constrains, does not appreciably quench the fluorescence of free AC.
Finally, it is interesting to note that the AC-amide hydrogen-bonded complexes
have slightly larger fluorescence quantum yields, f
, and longer lifetimes, t , than free AC. Therefore,
because f = kr/(kr+knr)
and t =1/(kr+knr), complexation
apparently hinders the non-radiative process, knr. In the singlet
manifold of the AC monomer, there exist two low-lying singlet excited states
that are analogous to the 1La and 1Lb
states of indole and other polyacenes [6]. In indoles, the 1Lb
state remains lower in the gas phase and non polar solvents, while the more
polar 1La state becomes the lowest energy excited singlet
state in polar solvents [16-18]. The fluorescence from 1La
is weaker than that from 1Lb. Coupling of both states
leads to an efficient nonradiative process involving the dissociation of the
NH pyrrolic group. Therefore environmental changes that induce decoupling of
these states would enhance both the quantum yield and fluorescence lifetime.
Since the close structural relation between AC and indole, it seems rather
likely that the 1La and 1Lb states
of AC behave in a similar way. Thus, we can tentatively suggest that the hydrogen-bonding
interaction between AC and the amides decouples the 1La
and 1Lb states of AC enhancing the quantum yield and fluorescence
lifetime.
In contrast to that, hydrogen-bonding interactions of AC with alcohols and
water are known to decrease the quantum yield and the fluorescence lifetime
of AC [8]. Waluk and coworkers have attributed this behaviour to the enhancement
of the internal conversion process caused by these hydrogen-bonding interactions
[8]. Alcohols and water are bifunctional donor-acceptor hydrogen-bonding molecules
that can interact with both the pyrrolic and the pyridinic nitrogen atoms of
AC. Therefore, we suspect that the interaction through the pyridinic nitrogen
atom is responsible of such a behaviour. In this case, a different mechanism
which deactivates the S1 state of AC could take place. In fact, Waluk
and coworkers have also considered the possibility of a hydrogen-bonding mediated
charge transfer process. We think that a systematic study of the hydrogen bonding
interactions of the pyridinic nitrogen atom of AC with selected hydrogen-bonded
donors could help to clarify this question. This study is now in progress in
our laboratory and it will be the subject of a forthcoming publication.
4. Acknowledgements
We gratefully thank financial support from Dirección General de Investigación
Científica y Técnica (PB98-1160) and Junta de Andalucía.
5. References
[1] R.A. Abramovitch, I.D. Spencer, The carbolines, Adv. Heterocycl. Chem.
3 (1964) 79-207.
[2] For a recent review see: J. Waluk, Conformational aspects of intra-
and intermolecular excited-state proton transfer, in: J. Waluk (Ed.), Conformational
analysis of molecules in excited states, Wiley-VCH, New York, 2000, chapter
2.
[3] C. Chang, N. Shabestary, A. El-Bayoumi, Excited-state double proton
transfer in 1-azacarbazole hydrogen-bonded dimers, Chem. Phys. Letters 75
(1980) 107-109.
[4] J. Sepiol, U.P. Wild, Excited-state double proton transfer in heterodimers
of 1-azacarbazole, Chem. Phys. Letters 93 (1982) 204-207.
[5] J. Waluk, A. Grabowska, B. Pakula, J. Sepiol, Viscosity vs. temperature
effects in excited-state double proton transfer. Comparison of 1-azacarbazole
with 7-azaindole, J. Phys. Chem. 88 (1984) 1160-1162.
[6] K. Fuke, T. Yabe, N. Chiba, T. Kohida, K. Kaya, Double-proton-transfer
reaction in the excited state of 1-azacarbazole dimer and 1-azacarbazole-7-azaindole
heterodimer studied in a supersonic jet, J. Phys. Chem. 90 (1986) 2309-2311.
[7] J. Waluk, J. Herbich, D. Oelkrug, S. Uhl, Excited-state double proton
transfer in the solid state: the dimers of 1-azacarbazole, J. Phys. Chem.
90 (1986) 3866-3868.
[8] J. Waluk, S.J. Komorowski, J. Herbich, Excited-state double proton
transfer in 1-azacarbazole-alcohol complexes, J. Phys. Chem. 90 (1986) 3868-3871.
[9] K. Fuke, K. Tsukamoto, F. Misaizu, K. Kaya, Picosecond measurements
of the vibrationally resolved proton-transfer rate of jet-cooled 1-azacarbazole
dimer, J. Chem. Phys. 95 (1991) 4074-4080.
[10] C. Chang, F. Yen, P. Chou, C. Wei, Excited-state double proton transfer
in 1-azacarbazole hydrogen bonded complexes, J. Chin. Chem. Soc. 43 (1996)
463-472.
[11] S. Mente, S.J.V. Frankland, L. Reynolds, M. Maroncelli, Evaluation
of classical potential functions for hydrogen bonding in 7-azaindole and
1-azacarbazole complexes, Chem. Phys. Letters 293 (1998) 515-522.
[12] S. Mente, M. Maroncelli, Solvation and the excited-state tautomerization
of 7-azaindole and 1-azacarbazole: computer simulation in water and alcohol
solvents, J. Phys. Chem. 102 (1998) 3860-3876.
[13] L. Stephenson, W.K. Warburton, Synthesis of some substituted a
-carbolines, J. Chem. Soc. (C) (1970) 1355-1364.
[14] Y. Marcus, The properties of organic liquids that are relevant to
their use as solvating solvents, Chem. Soc. Rev. (1993) 409-416.
[15] J.R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum
Press, New York, 2nd ed., 1999, chapter 4.
[16] J.W. Hager, D.R. Demmer, S.C. Wallace, Electronic spectra of jet-cooled
indoles: evidence for the 1La state, J. Phys. Chem.
91 (1987) 1375-1382.
[17] D.R. Demmer, G.W. Leach, E.A. Outhouse, J.W. Hager, S.C. Wallace,
Excited-state dynamics of jet-cooled substituted indoles: the role of polar
interactions in 1La-1Lb state
coupling, J. Phys. Chem. 94 (1990) 582-591.
[18] F. Gai, R.L. Rich, J.W. Petrich, Monophotonic ionization of 7-azaindole,
indole, and their derivatives and the role of overlapping excited states,
J. Am. Chem. Soc. 116 (1994) 735-746.